
Last Updated May 28, 2026
Speciation, diversity, and the tree of life examine how new species arise, how evolutionary branching generates the diversity of organisms on Earth, and how phylogenetic reasoning helps biology reconstruct the historical relationships among living and extinct lineages. Speciation is one of the central processes in biology because the richness of life does not emerge from variation within populations alone, but from the repeated splitting, divergence, persistence, hybridization, and extinction of lineages across deep time. Diversity is therefore not simply a count of kinds. It is the historical result of branching evolution under ecological, developmental, demographic, genomic, and geological constraint. The tree of life provides the framework through which common ancestry, lineage splitting, phylogenetic relationship, and evolutionary uncertainty are represented.
This article develops Speciation, Diversity, and the Tree of Life as a foundational article within the Biology knowledge series. It treats species formation not merely as a textbook definition of reproductive isolation, but as a dynamic process involving divergence, gene flow, ecological difference, developmental change, hybridization, lineage sorting, extinction, and uneven lineage persistence. It treats biodiversity not as a flat inventory, but as the accumulated outcome of branching history filtered by origination and loss. It treats phylogenetic trees not as decorative diagrams, but as explicit hypotheses about shared ancestry, branching order, divergence, uncertainty, and relationship.
Main Library
Publications
Article Map
Biology
Related Topic
Ecology
Related Topic
Earth Science
Related Topic
Environmental Science
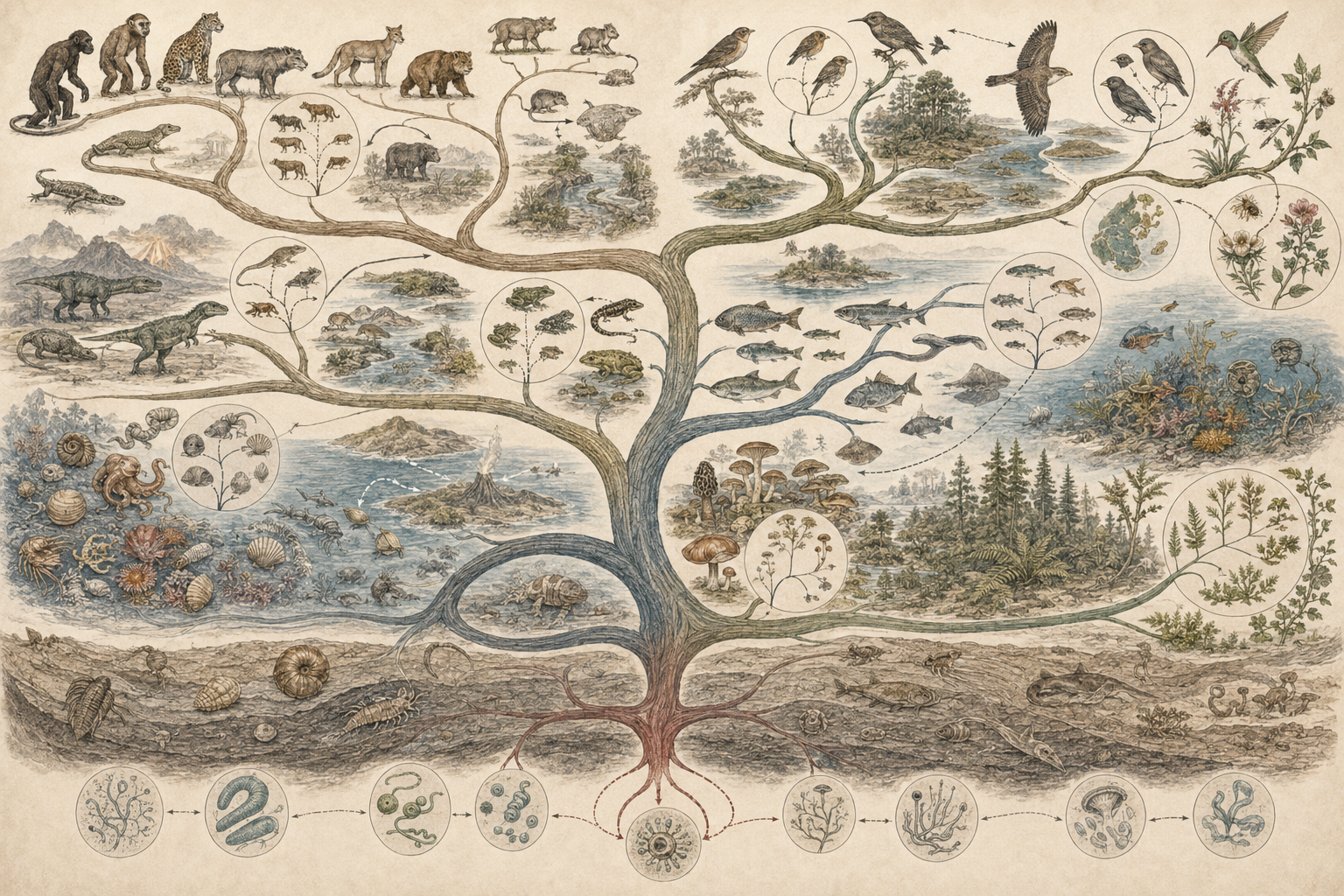
The article develops speciation, diversity, and the tree of life across species concepts, reproductive isolation, allopatric speciation, sympatric speciation, parapatric divergence, peripatric divergence, hybridization, introgression, phylogenetic reconstruction, incomplete lineage sorting, horizontal gene transfer, lineage richness, adaptive radiation, phylogenetic diversity, conservation risk, disease ecology, microbial evolution, plant diversification, marine and freshwater radiations, and computational phylogenetics.
The article also extends speciation biology into quantitative and computational analysis through allele-frequency divergence, \(F_{ST}\)-style population structure, sequence-distance calculation, Jukes-Cantor correction, distance-matrix construction, divergence-with-gene-flow simulation, birth-death diversification, lineage-through-time screening, R workflows, Python workflows, SQL provenance structures, and a linked full-stack GitHub repository containing Python, R, Julia, Fortran, Rust, Go, C, C++, SQL, notebooks, data files, validation notes, and reproducibility documentation.
What speciation and the tree of life are
Speciation is the evolutionary process through which new species arise. In classical terms, this often involves the evolution of barriers that reduce or prevent successful interbreeding between diverging populations. But for serious scientific work, speciation is broader than a single criterion. It includes the accumulation of lineage independence through reduced gene flow, ecological divergence, assortative mating, genomic incompatibility, developmental differentiation, chromosomal change, historical separation, and differential persistence through time.
The tree of life is the broader framework that situates speciation within common ancestry. As lineages diverge, branch, persist, hybridize, and go extinct, life’s history can be represented as a branching pattern of relatedness rather than an unordered collection of isolated forms. Tree thinking matters because biodiversity is genealogical. Organisms do not simply resemble one another; they inherit similarity and difference through branching descent.
Together, speciation and the tree of life explain why biodiversity exists in branching form. Life is diverse not simply because organisms vary, but because lineages repeatedly split, differentiate, persist, hybridize, and disappear. Diversity is therefore inseparable from branching history, and phylogenetic reasoning is inseparable from understanding biodiversity itself.
This also means that species are not merely names in a classification system. They are historical lineages with varying degrees of independence, coherence, reproductive continuity, ecological identity, and evolutionary trajectory. Some species boundaries are sharp. Others are porous, recent, hybridizing, or difficult to delimit. Speciation biology matters because it studies this transition from connected variation to historically structured diversity.
Species, divergence, and the origin of biological difference
Speciation matters because biodiversity is structured through divergence. Populations that once shared a common ancestral gene pool may become increasingly different in morphology, behavior, ecology, development, physiology, or genomic composition as gene flow declines and independent evolutionary trajectories emerge. These differences may begin modestly but accumulate through time into lineages that function as distinct species.
This process is historically important because it transforms variation within populations into larger-scale evolutionary diversity. A population can carry many differences, but species-level divergence requires that those differences become organized into lineages with some degree of long-term independence. Speciation is therefore the bridge between microevolutionary process and the larger branching structure of biodiversity.
Divergence also matters because it is not purely genetic in a narrow sense. It may involve ecological specialization, developmental timing, phenological separation, mate-recognition systems, host shifts, chromosomal change, habitat partitioning, physiological tolerance, or differences in reproductive strategy. Species originate when divergence becomes structured enough that lineages remain historically distinct rather than repeatedly collapsing back into one gene pool.
This makes speciation a multi-scale process. At one scale, allele frequencies diverge across populations. At another, genomes accumulate incompatibilities or locally adapted regions. At another, organisms differ in behavior, timing, ecology, or development. At still another, lineages become visible in phylogenetic history. Speciation is the gradual organization of difference into lineage independence.
Reproductive isolation and the logic of speciation
Reproductive isolation has traditionally been treated as one of the central criteria for speciation because it helps explain how diverging populations become evolutionarily independent. New species often form when opportunities for successful mating become reduced by ecological differences, behavioral differences, timing differences, mechanical differences, chromosomal differences, or genetic incompatibilities that affect hybrid viability or fertility.
This matters because it clarifies that speciation is not simply a matter of organisms looking different. Species boundaries often depend on whether lineages still exchange genes effectively, and if not, why not. The mechanisms that reduce exchange may arise gradually and in combination. Habitat divergence, sexual selection, chromosomal incompatibility, developmental mismatch, phenological separation, pollinator specificity, host shifts, and ecological specialization may all contribute to lineage separation.
At the same time, reproductive isolation is not always absolute, clean, or final. Some lineages hybridize after divergence, and species boundaries may remain porous in practice. For that reason, reproductive isolation is best understood not as a single all-or-nothing threshold, but as part of a broader process of lineage separation and partial independence distributed across genomes, ecologies, and time.
This distinction matters scientifically because reproductive isolation can be asymmetric, stage-specific, incomplete, or genome-region-specific. A pair of lineages may exchange genes at some loci while remaining distinct at others. They may produce hybrids that survive but have lower fertility. They may interbreed in rare contact zones but remain ecologically separate across most of their ranges. Speciation biology is therefore strongest when it treats isolation as a process, not as a simplistic yes-or-no switch.
Allopatric, sympatric, and other pathways of speciation
Biologists often distinguish different pathways of speciation based on geography and gene flow. Allopatric speciation occurs when populations become geographically separated, allowing divergence to accumulate under reduced gene exchange. Sympatric speciation refers to cases in which reproductive isolation emerges without complete geographic separation, often through ecological specialization, assortative mating, polyploidy, disruptive selection, or host shifts. Parapatric and peripatric patterns extend the framework further by emphasizing partial separation, edge populations, restricted contact zones, or founder effects.
These pathways matter because they show that speciation is not one uniform process. Geography often plays a major role, but ecological and genetic processes matter as well. Some lineages diverge because mountains, rivers, basins, islands, glaciers, deserts, currents, or habitat fragments split populations. Others diverge along ecological gradients, through reproductive timing, through host specialization, or because different mate preferences progressively reduce interbreeding.
Allopatric speciation has historically been central because physical separation provides a clear route to reduced gene flow. Yet allopatry is not sufficient by itself. Separated populations must also accumulate differences that persist when contact resumes, or remain geographically isolated long enough to become independently evolving lineages. Geographic separation can begin divergence, but speciation depends on what happens genetically, ecologically, developmentally, and reproductively after separation.
Sympatric and parapatric cases are especially important because they show that speciation can involve ecological differentiation even when populations remain near one another. Host-associated insects, plant polyploidy, habitat-specific mating, mate-preference divergence, and ecological specialization all show that lineage separation can be structured by resource use and reproductive behavior as well as by geography. The field is strongest when it treats speciation as a family of processes rather than a single recipe mechanically repeated across all life.
Hybridization, boundaries, and the instability of species limits
Species boundaries are not always permanent or tidy. Hybridization can blur lineages, transfer genetic material across species, or in some cases contribute to new evolutionary outcomes. Some speciation events remain incomplete. Some species exchange genes after divergence. Some lineages may even collapse back together under changing ecological conditions or under anthropogenic disturbance that disrupts previously maintained isolation.
This matters because biodiversity is not always best represented by perfectly sealed boxes. Species boundaries may be strong in some parts of the genome and weak in others. Hybrid zones may persist for long periods. Introgression can move adaptive alleles across species boundaries without eliminating lineage distinction. Conversely, shallow divergence may be enough for strong ecological or behavioral independence even when some gene flow continues.
Recognizing boundary instability does not weaken speciation theory. It strengthens it by making it more realistic. Diversity is historically branching, but not always perfectly clean-edged. Modern evolutionary biology gains explanatory power when it treats hybridization, partial isolation, reticulation, and lineage permeability as part of the real structure of speciation rather than as awkward exceptions.
This is especially relevant under human-driven environmental change. Habitat alteration can bring formerly separated lineages into contact, weaken ecological barriers, alter mating systems, or cause hybridization that threatens distinct lineages. In other cases, hybridization may introduce adaptive variation. Conservation biology therefore has to ask not only whether hybridization occurs, but what it means for lineage integrity, adaptive potential, ecological function, and long-term evolutionary history.
Diversity as the historical result of branching lineages
Biological diversity is best understood not merely as the existence of many species, but as the cumulative historical result of branching lineages shaped by speciation, extinction, adaptation, drift, ecological opportunity, and contingency. Diversity is genealogical before it is numerical. The number of species present at any one time reflects prior lineage splitting, prior lineage loss, and differential persistence across environments and clades.
This is why the tree of life is such a powerful framework. It captures both unity and plurality. All life is related, yet lineages have repeatedly branched into new forms under changing conditions. Diversity therefore reflects historical branching rather than a mere inventory of types. The pattern of life is genealogical, not simply classificatory.
Diversity is also unevenly distributed. Some clades contain immense species richness, while others are species-poor, relictual, or represented by only a few surviving lineages. Speciation, extinction, ecological release, developmental innovation, historical contingency, dispersal, and long-term stability all help shape these asymmetries. Understanding biodiversity therefore requires tracking both origination and loss rather than assuming present richness directly reflects evolutionary success in any simple sense.
This also matters for conservation. Losing a species is not equivalent to removing one interchangeable unit from a list. It may mean losing a long, distinctive branch of evolutionary history, a unique ecological function, a rare developmental trajectory, or a lineage with few close surviving relatives. Biodiversity has depth as well as breadth. Phylogenetic diversity helps make that depth visible.
The tree of life, common ancestry, and phylogenetic thinking
The tree of life rests on the principle of common ancestry. All living organisms are connected through ancestral branching, and nodes on a phylogenetic tree represent shared ancestors of descendant lineages. This matters because phylogenetic thinking changes how biology interprets resemblance. Similarity is not merely a matter of surface comparison. It may reflect shared ancestry, convergence, retained ancestral traits, parallel evolution, introgression, or later divergence.
Trees help sort these possibilities by asking how lineages are historically related rather than simply how much they resemble one another at first glance. That shift is foundational. It means that classification, comparative biology, trait evolution, and even functional inference become historical questions rather than purely descriptive ones.
The tree of life is therefore not just a visual summary. It is one of biology’s deepest explanatory frameworks, linking fossils, morphology, molecules, development, ecology, and classification into one historical structure. Without tree thinking, biodiversity appears as a scattered field of forms. With it, biodiversity becomes intelligible as branching descent.
Tree thinking also prevents evolutionary misunderstanding. Life is not arranged as a ladder from simple to complex, primitive to advanced, or lower to higher. It is a branching history of lineages, many of which persist, diversify, or go extinct under different conditions. Modern organisms are not ancestors of one another in a simple chain. They are relatives with shared ancestors. This distinction is essential for responsible evolutionary communication.
Phylogenetic trees as hypotheses of relationship
Phylogenetic trees are best understood as hypotheses about relationships, not as perfect photographs of the past. They are reconstructed from evidence rather than directly observed in full. Scientists compare homologous traits, fossil sequences, genomic patterns, molecular distances, developmental features, and other forms of data to infer branching history. Different datasets may refine, complicate, or sometimes challenge earlier hypotheses. Tree thinking is rigorous precisely because it remains evidence-based and revisable.
This is important because the tree of life is not a single kind of object. Some trees are inferred from morphology, some from a handful of genes, some from phylogenomics, some from species-level summaries, and some from fossilized character matrices. Their branch lengths may represent time, amount of change, or may be left unscaled altogether. Interpretation depends on model, data quality, sampling, character choice, taxonomic scope, and the assumptions used during inference.
Modern tree thinking has also been complicated by horizontal gene transfer, incomplete lineage sorting, hybridization, gene-tree/species-tree conflict, and reticulate evolution. These do not abolish phylogenetic reasoning. They make it more exacting. Biology is strongest when it treats trees as indispensable but methodologically complex representations of branching history.
This is especially important in genomics. Different genes may tell slightly different histories because they sort differently through ancestral populations, move laterally, introgress across species boundaries, or experience different selection pressures. A species tree is therefore not always identical to every gene tree. Strong phylogenetic inference requires acknowledging conflict rather than hiding it.
Speciation, development, and biological form
Speciation is not only a matter of reproductive boundaries or sequence divergence. It also interacts with development, morphology, and the production of biological form. Diverging lineages may differ in timing, patterning, signaling, body-plan expression, life-history scheduling, or developmental regulation, and those differences can contribute to ecological divergence or reproductive incompatibility.
This matters because species are not abstract genetic units alone. They are living forms produced through development. Development influences what variation becomes viable, what morphologies emerge, and what kinds of divergence can persist. Speciation therefore intersects naturally with developmental biology and evolutionary developmental biology, especially when lineage splitting involves altered developmental trajectories, life-history transitions, or regulatory architecture.
Biology is strongest here when it sees speciation as multi-scale at once: genomic, developmental, ecological, and historical. A species boundary may be maintained partly by gene-flow reduction, but also by developmental incompatibilities, phenological mismatch, ecological specialization, chromosomal structure, and divergent life-cycle organization.
Developmental change can also make speciation visible. Altered flowering time can separate plant populations. Shifted larval development can alter marine dispersal. Different mating structures can arise through developmental timing. Morphological divergence can reshape feeding, locomotion, reproduction, or habitat use. Speciation therefore often involves the interaction of lineage history with the developmental production of form and function.
Ecology, conservation, and systems risk
Speciation and phylogenetic diversity are deeply relevant to ecology and sustainability-adjacent biology because biodiversity is not just a present-day count of organisms. It is the result of long histories of divergence, adaptation, extinction, ecological interaction, and lineage persistence. Conservation therefore concerns not only species numbers but also lineage history, distinctiveness, function, and future evolutionary potential.
This is especially important in fragmented landscapes, rapidly changing climates, and systems under pollution, exploitation, invasive disruption, disease pressure, or hydrologic stress, where populations may be diverging, collapsing, hybridizing, or losing the conditions under which speciation and persistence occur. Conservation biology therefore faces a double problem: preventing immediate disappearance while also preserving the conditions for future lineage continuity and diversification.
For sustainability-oriented biology, this means the tree of life is not an abstract diagram. It is a record of irreplaceable evolutionary history. When lineages disappear, the loss is genealogical and functional, not merely numerical. Protecting biodiversity is also protecting branching history and the evolutionary possibility it carries.
Systems risk appears when lineages vanish faster than evolutionary processes can replace them, when fragmentation prevents gene flow needed for viability, when hybridization erodes distinctive lineages, or when climate change uncouples species from the ecological conditions under which they diversified. Speciation is slow relative to many human pressures. Conservation therefore often must protect the inherited results of deep time under conditions of rapid disturbance.
Marine, freshwater, soil, plant, and microbial relevance
Marine biology is deeply shaped by speciation and phylogenetic diversity. Oceans contain long histories of branching lineages, adaptive radiations, extinction events, and ecological partitioning across reefs, pelagic zones, benthic systems, estuaries, polar waters, and deep-sea habitats. Environmental gradients in temperature, salinity, oxygen, acidity, light, pressure, and depth can structure divergence and lineage persistence in marine organisms.
Freshwater systems also create strong conditions for divergence through drainage separation, lake basins, wetland mosaics, hydrologic fragmentation, and local adaptation. Many freshwater radiations illustrate how ecological heterogeneity and partial isolation can generate biodiversity through time. At the same time, freshwater systems are among the most vulnerable to fragmentation, pollution, invasive species, warming, altered flows, and habitat loss, making their phylogenetic and evolutionary histories especially important for conservation.
Plant science, agroecology, forestry, and restoration ecology show similar links among habitat structure, reproductive biology, dispersal, pollination, phenology, polyploidy, hybridization, and long-term lineage diversification. Plants can speciate through geographic isolation, ecological specialization, polyploidy, pollinator shifts, flowering-time divergence, and hybridization. This makes plant evolution particularly important for understanding both natural biodiversity and managed systems.
Soil biology and microbiology complicate tree thinking further because microbial evolution includes extensive lateral exchange alongside lineage divergence. This makes microbial history especially rich and sometimes less cleanly tree-like at fine genomic scales. Yet ancestry, branching, and diversification remain indispensable. Biology is strongest when it acknowledges both the power and the limits of strict tree representation across different domains of life.
Medical, biomedical, and disease ecology relevance
Speciation and phylogenetic relatedness matter in medicine and biomedicine because pathogens diverge, host lineages differ in susceptibility, model organisms are chosen partly on the basis of relatedness and conserved biology, and comparative genomics often depends on understanding branching history. Disease ecology extends these issues by showing how pathogen diversification, host shifts, hybridization, recombination, and lineage separation can alter transmission and virulence patterns.
This matters because the tree of life is not only about ancient natural history. It also shapes modern comparative reasoning. Closely related organisms may share important biological systems, while lineage divergence may explain differences in host range, immune interaction, tissue tropism, drug susceptibility, metabolism, or treatment response. Speciation therefore matters not only for taxonomy, but for how biology generalizes knowledge across organisms.
In biomedical and disease-ecological contexts, phylogenetic thinking connects present-day function to historical relationship. It helps explain why some pathogens jump hosts while others remain constrained, why model organisms can illuminate human biology, and why lineage context matters for interpreting virulence, resistance, and comparative physiology.
Speciation and lineage divergence also matter for surveillance. If a pathogen lineage splits into ecologically or clinically distinct lineages, public health interpretation changes. If recombination or reassortment blurs boundaries, tree-based inference must be combined with network-aware reasoning. Phylogenetics is therefore both a historical science and a practical tool for contemporary biological monitoring.
Phylogenomics, bioinformatics, and computational relevance
The tree of life has become even more powerful in the genomic era because lineage relationships can now be studied through large-scale sequence comparison as well as anatomy and fossils. Phylogenomics, comparative transcriptomics, model-based tree inference, divergence-time estimation, species-tree inference, gene-tree conflict analysis, and diversification-rate analysis all allow biology to link genome-scale evidence to branching history across vast taxonomic scope.
Bioinformatics is indispensable here because phylogenetics and phylogenomics depend on sequence alignment, model selection, distance estimation, branching inference, conflict analysis, support values, tree comparison, metadata integrity, and increasingly large genomic datasets. The modern tree of life is therefore both a biological theory and a computational achievement. It sits at the intersection of natural history, molecular biology, statistics, and algorithm design.
This makes speciation and tree thinking one of the strongest bridges between classical biology and data-rich contemporary science. For computational readers, the subject is especially useful because it combines inference under uncertainty, model dependence, branching structures, noisy historical signals, incomplete lineage sorting, gene-tree/species-tree conflict, reticulation, and biologically meaningful interpretation.
Reproducibility is especially important. Sequence selection, alignment quality, outgroup choice, model assumptions, taxon sampling, missing data, recombination, horizontal transfer, and convergence can all influence inferred relationships. A strong computational phylogenetic workflow therefore requires transparent provenance as well as biological judgment. A tree is never just a graphic. It is the output of a chain of assumptions and evidence.
Quantitative speciation biology: mathematics, R, and Python
Speciation and diversity are not only descriptive. They can also be approached quantitatively through population divergence, gene flow, branching models, phylogenetic distance, and lineage-splitting simulations. The aim of modeling is not to reduce species to numbers, but to make divergence, isolation, branching, and relatedness more explicit and analytically testable.
At the population-genetic level, divergence can begin with changes in allele frequencies. For a two-allele locus:
p+q=1
\]
Interpretation: At a biallelic locus, the two allele frequencies sum to one.
where \(p\) and \(q\) are allele frequencies. If two populations begin with the same frequencies and later diverge, part of speciation’s early process can be represented as accumulated population-level difference.
A simple absolute allele-frequency divergence between two populations can be written as:
\Delta p=|p_1-p_2|
\]
Interpretation: Absolute allele-frequency divergence measures how far apart two populations are at one locus.
Across many loci, average divergence can be summarized as:
\overline{\Delta p}=\frac{1}{L}\sum_{i=1}^{L}|p_{1i}-p_{2i}|
\]
Interpretation: Mean allele-frequency divergence summarizes population separation across many loci.
where \(L\) is the number of loci compared. This is useful because early lineage separation often becomes visible first as structured allele-frequency divergence before complete reproductive isolation is achieved.
Population differentiation is often summarized with an \(F_{ST}\)-style statistic:
F_{ST}=\frac{H_T-H_S}{H_T}
\]
Interpretation: \(F_{ST}\)-style summaries estimate how much genetic variation is partitioned among populations relative to total variation.
where \(H_T\) is total expected heterozygosity across pooled populations and \(H_S\) is mean expected heterozygosity within populations. Larger values imply stronger population structure. In real systems, interpretation depends on marker type, sampling scheme, demographic history, and statistical estimator, but the logic is useful: speciation often proceeds through increasing structure and decreasing effective gene exchange.
A basic measure of sequence divergence between equal-length sequences can be written as:
d=\frac{m}{L}
\]
Interpretation: Observed sequence distance is the fraction of aligned positions that differ.
where \(m\) is the number of differing positions and \(L\) is sequence length. A Jukes-Cantor correction gives:
d_{\mathrm{JC}}=-\frac{3}{4}\ln\left(1-\frac{4d}{3}\right)
\]
Interpretation: Jukes-Cantor correction adjusts observed distance for hidden substitutions under a simple nucleotide-substitution model.
This matters because raw mismatch proportions underestimate deeper divergence when multiple substitutions accumulate at the same sites.
At macroevolutionary scale, lineage richness reflects both speciation and extinction. A simple net diversification rate is:
r=\lambda-\mu
\]
Interpretation: Net diversification is the difference between speciation rate and extinction rate.
where \(\lambda\) is speciation rate and \(\mu\) is extinction rate. Under a simplified constant-rate approximation, expected lineage richness can be written as:
N(t)=N_0e^{rt}
\]
Interpretation: Lineage richness grows exponentially under a simplified constant net diversification rate.
This is analytically useful because biodiversity depends not only on how often new species arise, but on how often lineages are lost.
A simple lineage-through-time count can be represented as:
L(t)=\sum_{i=1}^{n} I_i(t)
\]
Interpretation: Lineage richness at time \(t\) can be represented as the sum of lineages present at that time.
where \(I_i(t)\) is an indicator for whether lineage \(i\) exists at time \(t\). This is useful because branching history is not only about final richness, but about the temporal accumulation and loss of lineages.
Variables, units, and speciation interpretation
Quantitative speciation biology depends on variables that connect population divergence, sequence distance, gene flow, population structure, diversification, lineage persistence, and biological interpretation. The table below summarizes several central quantities.
| Symbol or Term | Meaning | Typical Unit or Scale | Speciation Interpretation |
|---|---|---|---|
| \(p, q\) | Allele frequencies at a biallelic locus | fraction from 0 to 1 | Starting point for population-level divergence analysis |
| \(p_1, p_2\) | Allele frequencies in two populations | fraction from 0 to 1 | Population-specific allele frequencies under divergence |
| \(\Delta p\) | Absolute allele-frequency divergence | fractional difference | Simple measure of divergence between two populations at one locus |
| \(\overline{\Delta p}\) | Mean allele-frequency divergence across loci | average fractional difference | Genome-wide or multi-locus summary of population separation |
| \(L\) | Number of loci or sequence length depending on context | loci, nucleotides, base pairs, or sites | Number of positions compared in divergence analysis |
| \(H_T\) | Total expected heterozygosity | fraction from 0 to 1 | Diversity expected after pooling populations |
| \(H_S\) | Mean within-population heterozygosity | fraction from 0 to 1 | Average diversity within populations |
| \(F_{ST}\) | Population-differentiation statistic | dimensionless fraction-like statistic | Summary of structure among populations |
| \(m\) in sequence context | Mismatch count | count | Number of sequence positions that differ |
| \(d\) | Observed sequence distance | fraction from 0 to 1 | Raw proportion of aligned positions that differ |
| \(d_{\mathrm{JC}}\) | Jukes-Cantor corrected distance | substitutions per site under a simple model | Corrected divergence estimate accounting for hidden substitutions |
| \(\lambda\) | Speciation or origination rate | lineages per lineage per time | Rate at which new lineages arise |
| \(\mu\) | Extinction rate in diversification context | lineages per lineage per time | Rate at which lineages are lost |
| \(r\) | Net diversification rate | per time | Speciation rate minus extinction rate |
| \(N_0\) | Initial lineage richness | number of lineages | Starting richness in a diversification model |
| \(N(t)\) | Lineage richness at time \(t\) | number of lineages | Expected or observed number of lineages through time |
| \(I_i(t)\) | Lineage-presence indicator | 0 or 1 | Whether lineage \(i\) exists at time \(t\) |
| Gene flow | Exchange of genetic material among populations | migration rate, admixture fraction, or inferred exchange | Can slow divergence, homogenize lineages, or move adaptive alleles across boundaries |
| Introgression | Gene movement across species boundaries | locus-specific or genome-wide proportion | Indicates porous boundaries or hybridization after divergence |
| Incomplete lineage sorting | Persistence of ancestral polymorphism across speciation events | gene-tree/species-tree discordance | Explains why gene trees may differ from species trees |
The table shows why speciation quantities require context. An \(F_{ST}\)-style statistic, sequence distance, diversification rate, or lineage count becomes biologically meaningful only when linked to sampling design, organismal biology, gene flow, species concept, ecological setting, timescale, extinction risk, and analytical pipeline.
Worked example: population divergence, sequence distance, and lineage richness
Suppose two populations have allele frequencies \(p_1=0.72\) and \(p_2=0.41\) at a locus. Their absolute allele-frequency divergence is:
\Delta p=|0.72-0.41|=0.31
\]
Interpretation: The two populations differ by 0.31 in allele frequency at this locus.
This does not prove speciation by itself, but it indicates population-level genetic structure that could be part of a divergence process.
Now suppose total heterozygosity across two populations is \(H_T=0.50\), while mean within-population heterozygosity is \(H_S=0.38\). Then:
F_{ST}=\frac{H_T-H_S}{H_T}=\frac{0.50-0.38}{0.50}=0.24
\]
Interpretation: Under this simplified calculation, 24 percent of the modeled variation is partitioned among populations.
This is useful because speciation often begins with structured divergence among populations, though reproductive isolation and lineage history require additional evidence.
For sequence divergence, suppose two aligned sequences of length \(L=20\) differ at \(m=3\) positions:
d=\frac{3}{20}=0.15
\]
Interpretation: The observed proportion difference is 0.15.
A Jukes-Cantor correction gives:
d_{\mathrm{JC}}=-\frac{3}{4}\ln\left(1-\frac{4(0.15)}{3}\right)\approx0.168
\]
Interpretation: The corrected distance is larger than the observed difference because hidden substitutions may have occurred.
This is useful because phylogenetic inference often begins from observed differences but must account for the possibility that raw mismatch counts underestimate historical change.
At macroevolutionary scale, suppose a clade begins with \(N_0=12\) lineages, with speciation rate \(\lambda=0.09\) and extinction rate \(\mu=0.04\) per lineage per time unit. The net diversification rate is:
r=\lambda-\mu=0.09-0.04=0.05
\]
Interpretation: The simplified net diversification rate is 0.05 per time unit.
After \(t=25\) time units under a constant-rate approximation:
N(t)=12e^{0.05(25)}=12e^{1.25}\approx41.9
\]
Interpretation: The simplified expected lineage richness is approximately 42 lineages.
This matters because diversity depends on both lineage origination and lineage loss, not on speciation alone.
R and Python workflows
Computational modeling helps make speciation and phylogenetic reasoning explicit because lineage divergence is historical, stochastic, multi-locus, and often uncertain. Divergence-with-gene-flow simulations show how populations can separate while still exchanging alleles. \(F_{ST}\)-style summaries represent population structure. Sequence-distance workflows turn aligned sequences into distance matrices. Jukes-Cantor correction illustrates how molecular distance can be adjusted for hidden substitutions. Birth-death models represent the balance between speciation and extinction. Lineage-through-time summaries help analyze how diversity accumulates or collapses across time.
The selected examples below focus on compact, reusable workflows: multi-locus divergence with limited migration, \(F_{ST}\)-style structure, pairwise distance matrices, Jukes-Cantor correction, UPGMA-style clustering inputs, birth-death diversification, and lineage-richness screening. The GitHub repository extends the same logic into richer workflows for SQL provenance, reproducible data files, validation notes, notebooks, and multi-language scientific-computing examples.
The purpose is not to reduce speciation to code. The purpose is to make divergence and branching reasoning inspectable. A speciation or phylogenetic claim becomes stronger when allele frequencies, sequence data, population definitions, gene-flow assumptions, taxon sampling, model choices, lineage metadata, and analytical code are documented together.
R workflow: divergence, distance matrices, and birth-death diversification
R is useful for speciation and phylogenetic analysis because it supports simulation, statistical summaries, tabular workflows, distance matrices, clustering inputs, and reproducible reporting. The following workflow simulates two diverging populations across many loci under drift, weak selection, and limited migration; constructs a pairwise sequence-distance matrix; and simulates stochastic birth-death diversification.
# Speciation, Diversity, and the Tree of Life Workflow
#
# This workflow demonstrates three quantitative speciation tasks:
#
# 1. Simulate two diverging populations across many loci under drift,
# weak selection, and limited migration.
# 2. Build pairwise raw-distance and Jukes-Cantor distance matrices
# for aligned lineages.
# 3. Simulate simple birth-death diversification and lineage richness.
#
# These examples can be adapted for evolutionary biology, conservation
# genetics, phylogenetics, disease ecology, comparative genomics,
# freshwater radiations, marine population structure, and biodiversity science.
library(dplyr)
library(tidyr)
library(purrr)
library(stringr)
library(tibble)
# ------------------------------------------------------------
# 1. Multi-locus divergence with limited gene flow
# ------------------------------------------------------------
simulate_two_populations <- function(
generations = 250,
loci = 250,
N1 = 300,
N2 = 300,
m12 = 0.002,
m21 = 0.002,
sel_sd = 0.015,
seed = 42
) {
set.seed(seed)
p1 <- runif(loci, 0.2, 0.8)
p2 <- p1
s1 <- rnorm(loci, mean = 0, sd = sel_sd)
s2 <- -s1
records <- vector("list", generations + 1)
record_state <- function(gen, p1, p2) {
H1 <- 2 * p1 * (1 - p1)
H2 <- 2 * p2 * (1 - p2)
p_bar <- (p1 + p2) / 2
HT <- 2 * p_bar * (1 - p_bar)
HS <- (H1 + H2) / 2
fst <- ifelse(HT > 0, (HT - HS) / HT, 0)
tibble(
generation = gen,
locus = seq_along(p1),
p1 = p1,
p2 = p2,
delta_p = abs(p1 - p2),
H1 = H1,
H2 = H2,
HT = HT,
HS = HS,
fst = fst
)
}
records[[1]] <- record_state(0, p1, p2)
for (g in 1:generations) {
p1_sel <- p1 + s1 * p1 * (1 - p1)
p2_sel <- p2 + s2 * p2 * (1 - p2)
p1_sel <- pmin(pmax(p1_sel, 0), 1)
p2_sel <- pmin(pmax(p2_sel, 0), 1)
p1_mig <- (1 - m12) * p1_sel + m12 * p2_sel
p2_mig <- (1 - m21) * p2_sel + m21 * p1_sel
p1 <- rbinom(loci, 2 * N1, p1_mig) / (2 * N1)
p2 <- rbinom(loci, 2 * N2, p2_mig) / (2 * N2)
records[[g + 1]] <- record_state(g, p1, p2)
}
bind_rows(records)
}
divergence_sim <- simulate_two_populations()
summary_time <- divergence_sim %>%
group_by(generation) %>%
summarise(
mean_delta_p = mean(delta_p),
mean_fst = mean(fst),
max_fst = max(fst),
.groups = "drop"
)
outlier_loci <- divergence_sim %>%
filter(generation == max(generation)) %>%
arrange(desc(fst)) %>%
slice_head(n = 15)
# ------------------------------------------------------------
# 2. Pairwise sequence-distance matrix
# ------------------------------------------------------------
seqs <- c(
lineage_A = "ATGCTAGCTAACGGTACCTA",
lineage_B = "ATGCTGGCTATCGGTACCTA",
lineage_C = "ATGATGGCTATCGGTTCCTA",
lineage_D = "ATGCTAGTTAACGGAACCTG",
lineage_E = "ATGCTAGCTAACGGAACCTA"
)
distance_fun <- function(x, y) {
sx <- str_split(x, "", simplify = TRUE)
sy <- str_split(y, "", simplify = TRUE)
mismatches <- sum(sx != sy)
L <- length(sx)
p_distance <- mismatches / L
jukes_cantor <- ifelse(
p_distance >= 0.75,
NA_real_,
-(3 / 4) * log(1 - (4 / 3) * p_distance)
)
tibble(
mismatches = mismatches,
p_distance = p_distance,
jukes_cantor = jukes_cantor
)
}
pairs <- expand.grid(
taxon1 = names(seqs),
taxon2 = names(seqs),
stringsAsFactors = FALSE
) %>%
as_tibble() %>%
filter(taxon1 < taxon2) %>%
mutate(
res = map2(
taxon1,
taxon2,
~ distance_fun(seqs[[.x]], seqs[[.y]])
)
) %>%
unnest(res)
taxa <- names(seqs)
dist_mat <- matrix(
0,
length(taxa),
length(taxa),
dimnames = list(taxa, taxa)
)
for (i in seq_len(nrow(pairs))) {
a <- pairs$taxon1[i]
b <- pairs$taxon2[i]
dist_mat[a, b] <- pairs$jukes_cantor[i]
dist_mat[b, a] <- pairs$jukes_cantor[i]
}
hc <- hclust(as.dist(dist_mat), method = "average")
# ------------------------------------------------------------
# 3. Birth-death diversification screening
# ------------------------------------------------------------
simulate_birth_death <- function(
steps = 120,
N0 = 8,
lambda = 0.10,
mu = 0.06,
seed = NULL
) {
if (!is.null(seed)) {
set.seed(seed)
}
N <- numeric(steps + 1)
N[1] <- N0
for (t in 2:(steps + 1)) {
current <- N[t - 1]
births <- rpois(1, lambda = lambda * current)
deaths <- rpois(1, lambda = mu * current)
N[t] <- max(current + births - deaths, 0)
}
tibble(
time = 0:steps,
richness = N
)
}
params <- tibble(
scenario = c("net_positive", "near_equilibrium", "high_turnover"),
lambda = c(0.10, 0.07, 0.14),
mu = c(0.03, 0.06, 0.12)
)
runs <- params %>%
mutate(
sim = pmap(
list(lambda, mu),
~ simulate_birth_death(
steps = 120,
N0 = 8,
lambda = ..1,
mu = ..2,
seed = 42
)
)
) %>%
select(scenario, sim) %>%
unnest(sim)
diversification_summary <- runs %>%
group_by(scenario) %>%
summarise(
final_richness = last(richness),
peak_richness = max(richness),
.groups = "drop"
)
# ------------------------------------------------------------
# Print compact outputs
# ------------------------------------------------------------
print(
summary_time %>%
filter(generation %in% c(0, 50, 100, 150, 200, 250)) %>%
mutate(across(where(is.numeric), round, 4))
)
print(outlier_loci %>% mutate(across(where(is.numeric), round, 4)))
print(pairs %>% mutate(across(where(is.numeric), round, 4)))
print(round(dist_mat, 4))
print(hc)
print(diversification_summary)
print(runs %>% filter(time %in% c(0, 25, 50, 75, 100, 120)))This R workflow is useful because it models one of the real analytical questions in early speciation research: how divergence accumulates across many loci under limited migration, selection, and drift, and how genome-wide structure differs from locus-specific outliers. It then connects population divergence to sequence-distance reasoning and macroevolutionary diversification.
Python workflow: divergence with gene flow, distance matrices, and diversification screening
Python is useful for speciation and phylogenetic reasoning because it supports stochastic simulation, matrix operations, sequence comparison, pipeline design, data validation, and reproducible computation. The following workflow simulates divergence with limited migration, builds a pairwise sequence-distance matrix, and runs stochastic birth-death diversification screening.
"""
Speciation, Diversity, and the Tree of Life Workflow
This workflow demonstrates three quantitative speciation tasks:
1. Simulate divergence between two populations with limited migration,
drift, and weak opposing selection across many loci.
2. Build pairwise sequence-distance tables and Jukes-Cantor distance matrices.
3. Simulate stochastic birth-death diversification and summarize lineage risk.
The examples are compact, but the same structures can be extended to
evolutionary biology, conservation genetics, phylogenomics, disease ecology,
comparative genomics, marine and freshwater divergence, microbial evolution,
and biodiversity science.
"""
from __future__ import annotations
from itertools import combinations
import numpy as np
import pandas as pd
def simulate_divergence(
generations: int = 250,
loci: int = 300,
N1: int = 300,
N2: int = 300,
m12: float = 0.002,
m21: float = 0.002,
sel_sd: float = 0.015,
seed: int = 42,
) -> tuple[pd.DataFrame, pd.DataFrame, pd.DataFrame]:
"""
Simulate divergence between two populations with limited migration.
The model is intentionally compact:
- Initial allele frequencies are shared.
- Weak opposing locus-specific selection is applied.
- Limited migration connects the populations.
- Drift is applied through binomial sampling.
"""
rng = np.random.default_rng(seed)
p1 = rng.uniform(0.2, 0.8, size=loci)
p2 = p1.copy()
s1 = rng.normal(0.0, sel_sd, size=loci)
s2 = -s1
records = []
for generation in range(generations + 1):
H1 = 2.0 * p1 * (1.0 - p1)
H2 = 2.0 * p2 * (1.0 - p2)
pbar = (p1 + p2) / 2.0
HT = 2.0 * pbar * (1.0 - pbar)
HS = (H1 + H2) / 2.0
fst = np.where(HT > 0, (HT - HS) / HT, 0.0)
records.append(
pd.DataFrame(
{
"generation": generation,
"locus": np.arange(loci),
"p1": p1,
"p2": p2,
"delta_p": np.abs(p1 - p2),
"fst": fst,
}
)
)
if generation == generations:
break
p1_sel = np.clip(p1 + s1 * p1 * (1.0 - p1), 0.0, 1.0)
p2_sel = np.clip(p2 + s2 * p2 * (1.0 - p2), 0.0, 1.0)
p1_mig = (1.0 - m12) * p1_sel + m12 * p2_sel
p2_mig = (1.0 - m21) * p2_sel + m21 * p1_sel
p1 = rng.binomial(2 * N1, p1_mig) / (2.0 * N1)
p2 = rng.binomial(2 * N2, p2_mig) / (2.0 * N2)
df = pd.concat(records, ignore_index=True)
summary = (
df.groupby("generation")
.agg(
mean_delta_p=("delta_p", "mean"),
mean_fst=("fst", "mean"),
max_fst=("fst", "max"),
)
.reset_index()
)
final_generation = df["generation"].max()
outliers = (
df[df["generation"] == final_generation]
.sort_values("fst", ascending=False)
.head(20)
)
return df, summary, outliers
def pairwise_sequence_distance() -> tuple[pd.DataFrame, pd.DataFrame]:
"""
Build a pairwise sequence-distance table and Jukes-Cantor distance matrix.
"""
sequences = {
"lineage_A": "ATGCTAGCTAACGGTACCTA",
"lineage_B": "ATGCTGGCTATCGGTACCTA",
"lineage_C": "ATGATGGCTATCGGTTCCTA",
"lineage_D": "ATGCTAGTTAACGGAACCTG",
"lineage_E": "ATGCTAGCTAACGGAACCTA",
}
def distance(seq1: str, seq2: str) -> tuple[int, float, float]:
if len(seq1) != len(seq2):
raise ValueError("Sequences must be equal length for this compact example.")
mismatches = sum(a != b for a, b in zip(seq1, seq2))
length = len(seq1)
p_distance = mismatches / length
if p_distance >= 0.75:
jukes_cantor = np.nan
else:
jukes_cantor = -(3.0 / 4.0) * np.log(
1.0 - (4.0 / 3.0) * p_distance
)
return mismatches, p_distance, jukes_cantor
rows = []
for lineage_a, lineage_b in combinations(sequences.keys(), 2):
mismatches, p_distance, jukes_cantor = distance(
sequences[lineage_a],
sequences[lineage_b],
)
rows.append(
{
"taxon_1": lineage_a,
"taxon_2": lineage_b,
"mismatches": mismatches,
"p_distance": p_distance,
"jukes_cantor": jukes_cantor,
}
)
distance_df = pd.DataFrame(rows)
taxa = list(sequences.keys())
matrix = pd.DataFrame(
np.zeros((len(taxa), len(taxa))),
index=taxa,
columns=taxa,
)
for _, row in distance_df.iterrows():
matrix.loc[row["taxon_1"], row["taxon_2"]] = row["jukes_cantor"]
matrix.loc[row["taxon_2"], row["taxon_1"]] = row["jukes_cantor"]
return distance_df, matrix
def birth_death_simulation(
time_steps: int = 120,
N0: int = 8,
lambda_rate: float = 0.10,
mu_rate: float = 0.06,
n_iter: int = 1000,
seed: int = 7,
) -> tuple[pd.DataFrame, pd.DataFrame]:
"""
Simulate stochastic lineage richness under birth-death logic.
"""
rng = np.random.default_rng(seed)
finals = []
peaks = []
extinct_by_end = []
for _ in range(n_iter):
richness = N0
peak = richness
for _ in range(time_steps):
births = rng.poisson(lambda_rate * richness)
deaths = rng.poisson(mu_rate * richness)
richness = max(richness + births - deaths, 0)
peak = max(peak, richness)
if richness == 0:
break
finals.append(richness)
peaks.append(peak)
extinct_by_end.append(richness == 0)
runs = pd.DataFrame(
{
"final_richness": finals,
"peak_richness": peaks,
"extinct_by_end": extinct_by_end,
}
)
summary = pd.DataFrame(
{
"extinction_probability": [runs["extinct_by_end"].mean()],
"mean_final_richness": [runs["final_richness"].mean()],
"median_final_richness": [runs["final_richness"].median()],
"mean_peak_richness": [runs["peak_richness"].mean()],
}
)
return runs, summary
def lineage_condition_score() -> pd.DataFrame:
"""
Build a compact lineage-condition screen for conservation and systems analysis.
"""
systems = pd.DataFrame(
{
"system": [
"fragmented_freshwater_radiation",
"hybridizing_plant_complex",
"marine_larval_dispersal_network",
"pathogen_species_complex",
"relict_forest_lineage",
],
"lineage_distinctiveness": [0.82, 0.55, 0.44, 0.68, 0.91],
"gene_flow_complexity": [0.30, 0.86, 0.72, 0.64, 0.18],
"habitat_fragmentation": [0.88, 0.48, 0.35, 0.42, 0.76],
"extinction_risk": [0.74, 0.38, 0.30, 0.52, 0.81],
"phylogenetic_information_quality": [0.62, 0.58, 0.70, 0.76, 0.49],
}
)
systems["conservation_priority_score"] = (
0.35 * systems["lineage_distinctiveness"]
+ 0.30 * systems["extinction_risk"]
+ 0.20 * systems["habitat_fragmentation"]
+ 0.15 * (1.0 - systems["phylogenetic_information_quality"])
)
systems["tree_inference_complexity_score"] = (
0.40 * systems["gene_flow_complexity"]
+ 0.30 * (1.0 - systems["phylogenetic_information_quality"])
+ 0.30 * systems["habitat_fragmentation"]
)
return systems.sort_values("conservation_priority_score", ascending=False)
def main() -> None:
"""
Run compact speciation and tree-of-life workflows.
"""
_, divergence_summary, divergence_outliers = simulate_divergence()
distance_df, distance_matrix = pairwise_sequence_distance()
diversification_runs, diversification_summary = birth_death_simulation()
condition_score = lineage_condition_score()
print("Divergence-with-gene-flow summary:")
print(divergence_summary.head(15).round(4).to_string(index=False))
print(divergence_summary.tail(15).round(4).to_string(index=False))
print("\nHigh-differentiation loci at final generation:")
print(divergence_outliers.round(4).to_string(index=False))
print("\nPairwise sequence-distance table:")
print(distance_df.round(4).to_string(index=False))
print("\nJukes-Cantor distance matrix:")
print(distance_matrix.round(4).to_string())
print("\nBirth-death diversification summary:")
print(diversification_summary.round(4).to_string(index=False))
print(diversification_runs.describe().round(4).to_string())
print("\nLineage condition score:")
print(condition_score.round(4).to_string(index=False))
if __name__ == "__main__":
main()This Python workflow is useful because it produces something closer to a real speciation-analysis scaffold: multi-locus divergence, genome-wide structure, high-differentiation loci under ongoing but limited migration, distance-matrix construction, stochastic diversification, and applied lineage-condition screening. It provides a compact bridge between population divergence, phylogenetic inference, and conservation-oriented biodiversity analysis.
GitHub repository
The article body includes compact R and Python examples so the biological and scientific argument remains readable. The full repository expands those examples into a broader computational speciation and phylogenetics workflow, including divergence-with-gene-flow simulation, \(F_{ST}\)-style population-structure screening, pairwise sequence distances, Jukes-Cantor correction, distance-matrix construction, UPGMA-style clustering inputs, birth-death diversification, lineage-through-time screening, lineage-condition scoring, SQL provenance structures, reproducible data files, validation notes, and full-stack scientific-computing examples across Python, R, Julia, Fortran, Rust, Go, C, C++, SQL, and notebooks.
Limits, complexity, and modern tree thinking
Speciation and the tree of life are foundational, but neither should be oversimplified. Species are sometimes difficult to delimit cleanly. Trees are hypotheses rather than perfect final maps. Horizontal gene transfer, hybridization, incomplete lineage sorting, reticulation, introgression, gene-tree/species-tree conflict, and uneven rates of diversification can complicate branching history substantially. The strongest strict version of a universal perfectly tree-like history is too simple for the full complexity of life, especially in microbial evolution. But ancestry, divergence, and branching remain indispensable.
This is not a weakness of evolutionary biology. It is part of its maturity. Modern biology is strongest when it treats lineages, species, and trees as historically real but methodologically complex. The world is genealogical, but not always neat. Tree thinking remains powerful precisely because it is revisable, data-rich, and able to incorporate conflict and uncertainty rather than pretending those do not exist.
Models are useful because they clarify assumptions, expose mechanisms, and make comparison possible. But \(F_{ST}\) is not speciation by itself, a distance matrix is not a complete phylogeny, and a birth-death simulation is not the whole tree of life. Quantitative tools are strongest when they support biological interpretation rather than replacing it.
In that sense, speciation and phylogenetic reasoning exemplify modern biology itself: historical, evidence-rich, computationally refined, and resistant to naive simplification. Their strength lies not in pretending that species boundaries are always simple, but in explaining how lineage structure becomes real under conditions of divergence, gene flow, isolation, extinction, and deep time.
This caution matters in public and scientific communication alike. Species are not arbitrary labels, but they are also not always perfectly sealed categories. Trees are not decorative ladders of progress, but they are also not immune to uncertainty. Responsible tree thinking requires both historical confidence and methodological humility.
Why this matters for scientific work
For working scientists, speciation and phylogenetic thinking matter because many biological questions are misframed if species are treated as static units and relatedness as background detail. A conservation problem may hinge on whether populations represent distinct lineages or fragmented parts of one species. A disease problem may depend on host shifts or pathogen diversification. A restoration problem may turn on preserving local genetic structure or preventing lineage collapse through hybridization. A comparative study may fail if phylogenetic non-independence is ignored.
This means speciation and tree thinking should often be treated as explanatory infrastructure rather than as specialized theory. Ecologists need them to understand diversification, coexistence, and lineage assembly. Conservation scientists need them because biodiversity loss is also phylogenetic loss. Microbiologists need them because microbial evolution mixes vertical descent with lateral exchange. Plant scientists need them because polyploidy, hybridization, pollination, and phenology can shape lineage divergence. Biomedical scientists need them because historical relatedness structures comparative inference. Computational biologists need them because branching and reticulation are central data problems as well as biological realities.
The scientific importance of speciation lies partly in this breadth. It is one of the main ways biology explains how diversity becomes possible, how lineage history is reconstructed, and how living difference is organized through ancestry.
Speciation and tree thinking are also practically actionable. Divergence can be measured. Sequence distances can be calculated. Distance matrices can be constructed. Lineage richness can be modeled. Hybridization can be detected. Phylogenetic diversity can inform conservation. Pathogen lineages can be tracked. These tools connect evolutionary theory to conservation, disease ecology, comparative biology, genomics, restoration ecology, medicine, and biodiversity science.
Conclusion
Speciation, diversity, and the tree of life show that biodiversity is historically produced through branching lineages rather than merely assembled as a static inventory of forms. New species arise as populations diverge, reproductive barriers evolve, ecological and developmental differences accumulate, and lineages become historically distinct. The tree of life provides the conceptual and evidentiary framework through which those relationships are understood.
To understand speciation is therefore to understand how diversity becomes possible. To understand the tree of life is to understand how that diversity remains connected through common ancestry. Together, these ideas explain why life is both unified and plural, historically related and ecologically differentiated. That is why speciation and phylogenetic thinking remain central not only to evolutionary biology, but also to ecology, conservation, microbiology, plant science, marine and freshwater biology, disease ecology, medicine, biotechnology, and computational biology.
Speciation is thus more than the origin of species in a narrow sense. It is one of the principal ways biology explains the branching structure of life itself. Modern quantitative and computational workflows deepen that understanding by making divergence, gene flow, sequence distance, population structure, diversification, and phylogenetic provenance more transparent, reproducible, and scientifically interpretable.
Related articles
- Biology
- Evolution and the History of Life
- Natural Selection, Adaptation, and Fitness
- Population Genetics and the Mathematics of Inheritance
- Mutation, Variation, and the Sources of Novelty
- Genomics and the Expansion of Biological Knowledge
- Microevolution, Macroevolution, and Deep Time
- Extinction, Contingency, and Biological Transformation
- Biodiversity and the Structure of Living Systems
- Ecology and the Interdependence of Life
- Coevolution, Symbiosis, and the Dynamics of Mutual Change
- Population Dynamics and Ecological Modeling
Further reading
- Coyne, J.A. and Orr, H.A. (2004) Speciation. Sunderland, MA: Sinauer Associates. Bibliographic information available at: https://rcin.org.pl/dlibra/publication/159817/edition/141269?language=en
- Felsenstein, J. (2004) Inferring Phylogenies. Sunderland, MA: Sinauer Associates. Publisher information available at: https://global.oup.com/academic/product/inferring-phylogenies-9780878931774
- Futuyma, D.J. and Kirkpatrick, M. (2017) Evolution. 4th edn. Sunderland, MA: Sinauer Associates. Publisher information available at: https://global.oup.com/academic/product/evolution-9781605357409
- Hedges, S.B., Marin, J., Suleski, M., Paymer, M. and Kumar, S. (2015) ‘Tree of life reveals clock-like speciation and diversification’, Molecular Biology and Evolution, 32(4), pp. 835–845. Available at: https://doi.org/10.1093/molbev/msv037
- Maddison, W.P. and Knowles, L.L. (2006) ‘Inferring phylogeny despite incomplete lineage sorting’, Systematic Biology, 55(1), pp. 21–30. Available at: https://doi.org/10.1080/10635150500354928
- O’Malley, M.A., Koonin, E.V. and Martin, W. (2011) ‘How stands the tree of life a century and a half after The Origin?’, Biology Direct, 6, 32. Available at: https://doi.org/10.1186/1745-6150-6-32
- Presgraves, D.C. (2002) ‘Patterns of postzygotic isolation in Lepidoptera’, Evolution, 56(6), pp. 1168–1183. Available at: https://doi.org/10.1111/j.0014-3820.2002.tb01430.x
- Scholl, J.P. and Wiens, J.J. (2016) ‘Diversification rates and species richness across the tree of life’, Proceedings of the Royal Society B, 283, 20161334. Available at: https://doi.org/10.1098/rspb.2016.1334
- Templeton, A.R. (2001) ‘Using phylogeographic analyses of gene trees to test species status and processes’, Molecular Ecology, 10(3), pp. 779–791. Available at: https://doi.org/10.1046/j.1365-294x.2001.01199.x
- Wakeley, J. (2008) Coalescent Theory: An Introduction. Greenwood Village, CO: Roberts and Company. Bibliographic information available at: https://search.worldcat.org/title/184827993
References
- Coyne, J.A. and Orr, H.A. (2004) Speciation. Sunderland, MA: Sinauer Associates. Bibliographic information available at: https://rcin.org.pl/dlibra/publication/159817/edition/141269?language=en
- Felsenstein, J. (2004) Inferring Phylogenies. Sunderland, MA: Sinauer Associates. Publisher information available at: https://global.oup.com/academic/product/inferring-phylogenies-9780878931774
- Futuyma, D.J. and Kirkpatrick, M. (2017) Evolution. 4th edn. Sunderland, MA: Sinauer Associates. Publisher information available at: https://global.oup.com/academic/product/evolution-9781605357409
- Hedges, S.B., Marin, J., Suleski, M., Paymer, M. and Kumar, S. (2015) ‘Tree of life reveals clock-like speciation and diversification’, Molecular Biology and Evolution, 32(4), pp. 835–845. Available at: https://doi.org/10.1093/molbev/msv037
- Maddison, W.P. and Knowles, L.L. (2006) ‘Inferring phylogeny despite incomplete lineage sorting’, Systematic Biology, 55(1), pp. 21–30. Available at: https://doi.org/10.1080/10635150500354928
- O’Malley, M.A., Koonin, E.V. and Martin, W. (2011) ‘How stands the tree of life a century and a half after The Origin?’, Biology Direct, 6, 32. Available at: https://doi.org/10.1186/1745-6150-6-32
- Presgraves, D.C. (2002) ‘Patterns of postzygotic isolation in Lepidoptera’, Evolution, 56(6), pp. 1168–1183. Available at: https://doi.org/10.1111/j.0014-3820.2002.tb01430.x
- Scholl, J.P. and Wiens, J.J. (2016) ‘Diversification rates and species richness across the tree of life’, Proceedings of the Royal Society B, 283, 20161334. Available at: https://doi.org/10.1098/rspb.2016.1334
- Templeton, A.R. (2001) ‘Using phylogeographic analyses of gene trees to test species status and processes’, Molecular Ecology, 10(3), pp. 779–791. Available at: https://doi.org/10.1046/j.1365-294x.2001.01199.x
- Wakeley, J. (2008) Coalescent Theory: An Introduction. Greenwood Village, CO: Roberts and Company. Bibliographic information available at: https://search.worldcat.org/title/184827993