
Last Updated May 28, 2026
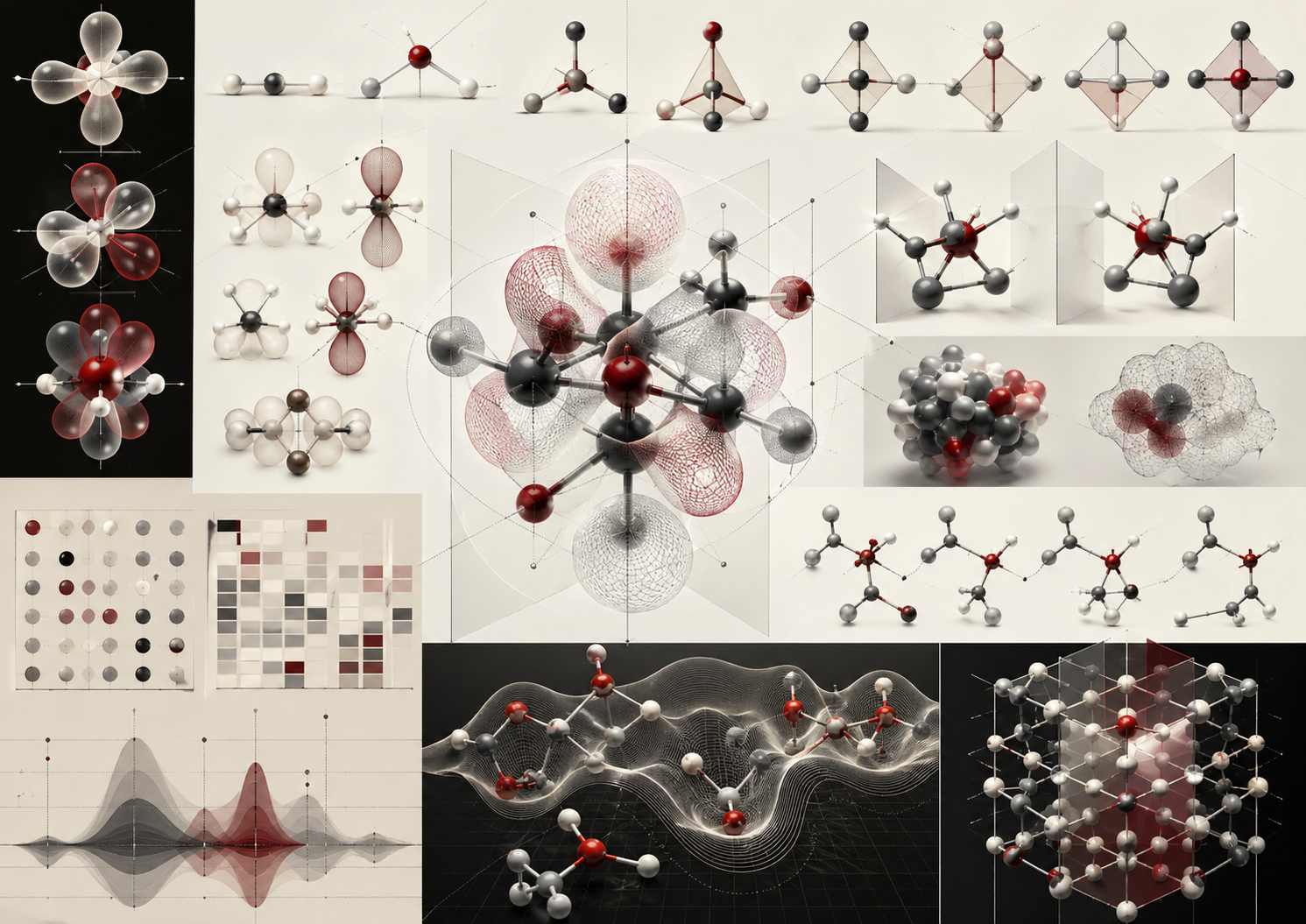
Molecular geometry is the spatial form of chemical bonding. A formula tells which atoms are present, and a connectivity diagram tells which atoms are joined, but molecular geometry tells how a molecule exists in three-dimensional space. Bond lengths, bond angles, torsion angles, stereochemistry, symmetry, conformations, electron-pair arrangements, orbital overlap, molecular surfaces, and crystal packing all shape the behavior of chemical systems.
The central thesis of this article is that molecular structure is not merely a drawing. It is an evidence-based, model-dependent, three-dimensional claim about matter. Structural chemistry requires symbolic formulas, experimental measurements, computational models, visual representations, and mathematical discipline. Good molecular reasoning knows the difference between a Lewis structure, a ball-and-stick model, an electron-density surface, a conformational ensemble, a crystallographic structure, and a computationally optimized geometry.
Molecular structure is central because chemical properties depend on spatial organization. Water is bent, carbon dioxide is linear, methane is tetrahedral, ammonia is trigonal pyramidal, benzene is planar and delocalized, proteins fold into complex three-dimensional structures, and crystalline materials organize molecular or ionic units into repeating lattices. Shape influences polarity, reactivity, spectroscopy, biological recognition, solubility, boiling point, crystal packing, catalysis, chirality, materials performance, and environmental fate.
Main Library
Publications
Article Map
Chemistry
Related Topic
Physics
Related Topic
Mathematical Modeling
Related Topic
Data Systems & Analytics
Why Molecular Geometry Matters
Molecular geometry matters because molecules are three-dimensional objects. Their behavior depends not only on which atoms are present, but on how those atoms are arranged. A molecule’s geometry influences electron distribution, bond polarity, molecular dipole, intermolecular forces, orbital overlap, reactivity, biological recognition, crystal packing, spectroscopic transitions, and materials behavior.
Water and carbon dioxide show the importance of geometry in a simple way. Both contain polar bonds. Water is bent and has a net molecular dipole. Carbon dioxide is linear and symmetrical, so its bond dipoles cancel. The difference in shape helps explain differences in physical behavior, environmental role, spectroscopy, and intermolecular interactions.
Geometry also shapes biological and medicinal chemistry. A drug molecule may bind to a protein because its three-dimensional shape complements a binding pocket. An enzyme may distinguish between stereoisomers. A protein’s function depends on its folded structure. DNA base pairing depends on geometry, hydrogen bonding, and stacking. Molecular structure is therefore not decorative; it is functional.
In materials chemistry, geometry controls porosity, mechanical strength, optical behavior, conductivity, catalytic surface exposure, and crystal packing. In environmental chemistry, molecular structure affects volatility, persistence, sorption, degradation, speciation, and bioavailability.
Geometry is also central to measurement. Spectroscopy, crystallography, diffraction, microscopy, computational chemistry, and structural databases all depend on spatial evidence. A chemical structure is not only an illustration; it is a claim about positions, distances, angles, symmetry, surfaces, and uncertainty.
Chemistry uses formulas, but molecules act through structure. To understand chemical behavior, chemists must understand matter as organized space.
Structure Beyond the Chemical Formula
A molecular formula gives composition. It does not fully define structure. The formula \(C_2H_6O\) can represent ethanol or dimethyl ether. These compounds contain the same numbers of carbon, hydrogen, and oxygen atoms, but their connectivity differs. Because their structures differ, their boiling points, reactivity, polarity, solubility, odor, and biological effects differ.
Connectivity is one structural layer. Three-dimensional geometry is another. Stereoisomers can have the same formula and connectivity but different spatial arrangement. Conformers can have the same connectivity and stereochemistry but differ by rotation around bonds. Crystalline phases can have the same molecular composition but different packing. Polymorphs can differ in solubility, stability, density, melting point, and mechanical behavior.
A structural description may include:
- molecular formula;
- connectivity;
- bond order;
- bond lengths;
- bond angles;
- torsion angles;
- stereochemistry;
- conformation;
- symmetry;
- electron density;
- molecular surface;
- crystal packing;
- dynamic motion;
- experimental uncertainty;
- model assumptions.
This hierarchy is important because no single representation captures all structure. A Lewis structure is useful for valence reasoning. A ball-and-stick model is useful for connectivity and geometry. A space-filling model is useful for steric shape. An electrostatic surface is useful for charge distribution. An electron-density map is useful for quantum and crystallographic evidence. A conformational ensemble is useful for flexible molecules. A crystal structure is useful for solid-state arrangement.
Molecular structure is therefore a layered scientific representation. It is strongest when the representation matches the question being asked.
Coordinates, Bond Lengths, and Bond Angles
A molecular geometry can be represented by atomic coordinates. Each atom is assigned a position in three-dimensional space:
\mathbf{r}_i = (x_i, y_i, z_i)
\]
Interpretation: The vector \(\mathbf{r}_i\) gives the spatial position of atom \(i\).
The distance between two atoms \(i\) and \(j\) is:
d_{ij} = \|\mathbf{r}_i – \mathbf{r}_j\|
\]
Interpretation: Bond distance or interatomic separation is the norm of the difference between two coordinate vectors.
In coordinate form:
d_{ij} = \sqrt{(x_i-x_j)^2+(y_i-y_j)^2+(z_i-z_j)^2}
\]
Interpretation: The three-dimensional distance formula calculates separation from Cartesian coordinates.
Bond angles are calculated from vectors. If atom \(B\) is the central atom in angle \(A-B-C\), the vectors are:
\mathbf{u} = \mathbf{r}_A – \mathbf{r}_B
\]
Interpretation: \(\mathbf{u}\) points from central atom \(B\) toward atom \(A\).
\mathbf{v} = \mathbf{r}_C – \mathbf{r}_B
\]
Interpretation: \(\mathbf{v}\) points from central atom \(B\) toward atom \(C\).
The angle is obtained from the dot product:
\cos\theta = \frac{\mathbf{u}\cdot\mathbf{v}}{\|\mathbf{u}\|\|\mathbf{v}\|}
\]
Interpretation: The dot product relates the angle between two bond vectors to their magnitudes and orientation.
These calculations are not merely mathematical exercises. Bond lengths and angles are used in crystallography, spectroscopy, molecular modeling, conformational analysis, protein structure, materials science, reaction mechanism studies, structural databases, and computational validation. They provide a quantitative language for shape.
The same molecule may also have different geometries under different conditions. Gas-phase geometry, solution structure, crystal structure, protein-bound structure, and computationally optimized structure may not be identical. Molecular geometry is therefore a measured or modeled structure under specified assumptions.
For researchers, coordinates are not just visualization data. They are the numerical basis of structural chemistry.
Torsion Angles, Conformation, and Flexibility
A torsion angle, or dihedral angle, describes rotation around a bond. It is defined by four atoms \(A-B-C-D\) and measures the angle between the plane formed by \(A-B-C\) and the plane formed by \(B-C-D\). Torsion angles are crucial in organic chemistry, biochemistry, polymer chemistry, coordination chemistry, and molecular modeling.
Conformation refers to spatial arrangements that can interconvert through rotation around single bonds without breaking bonds. Ethane has staggered and eclipsed conformations. Butane has anti and gauche conformations. Cyclohexane has chair, boat, and twist-boat conformations. Proteins have backbone torsion angles that shape secondary structure. Nucleic acids have sugar puckers and backbone conformations that affect structure.
Flexible molecules are not described by one geometry alone. They exist as ensembles of conformations, with populations depending on energy, temperature, solvent, steric effects, electronic effects, intramolecular interactions, and intermolecular interactions. A single static drawing may hide this dynamic reality.
Conformational analysis also distinguishes local minima from transition pathways. A molecule may prefer one conformer at low temperature, access several conformers at room temperature, or adopt a different conformation when bound to a receptor, packed in a crystal, dissolved in a solvent, or adsorbed on a surface.
This matters because reactivity and recognition often depend on accessible conformations. A reactive group may be present but geometrically misaligned. A binding ligand may need to adopt a rare but biologically relevant conformation. A polymer’s material behavior may depend on rotational freedom along its backbone.
Conformational analysis therefore connects molecular structure to thermodynamics, kinetics, spectroscopy, and biological function. Structure is not always a frozen object. Often it is a landscape.
VSEPR and Electron-Domain Geometry
Valence-shell electron-pair repulsion theory, or VSEPR, is a useful model for predicting molecular geometry from electron domains around a central atom. Electron domains include bonding pairs and lone pairs. The central idea is that electron domains tend to arrange themselves to reduce repulsion.
VSEPR distinguishes electron-domain geometry from molecular geometry. Electron-domain geometry includes both bonding pairs and lone pairs. Molecular geometry describes the arrangement of atoms. For methane, the electron-domain geometry and molecular geometry are both tetrahedral. For ammonia, the electron-domain geometry is tetrahedral, but the molecular geometry is trigonal pyramidal because one domain is a lone pair. For water, the electron-domain geometry is tetrahedral, but the molecular geometry is bent because two domains are lone pairs.
VSEPR is powerful because it connects valence electron structure to molecular shape. It gives students and chemists a practical way to predict geometries such as linear, trigonal planar, tetrahedral, trigonal bipyramidal, and octahedral.
However, VSEPR is not a complete theory of molecular structure. It is less reliable for many transition-metal complexes, delocalized systems, heavy-element compounds, hypervalent descriptions, multicenter bonding, and subtle energetic differences. It should be treated as an introductory structural model, not as the final explanation of geometry.
VSEPR also does not replace evidence. A predicted geometry should be tested or refined through spectroscopy, diffraction, computation, or comparison with reliable structural data when precision matters.
For researchers, VSEPR is useful because it gives an accessible first model. But structural chemistry becomes stronger when VSEPR is connected to orbital theory, electron density, measurement, and computational evidence.
Common Molecular Geometries
Many molecular geometries recur throughout chemistry. These geometries provide a shared structural vocabulary.
- Linear: atoms arranged in a straight line, as in carbon dioxide.
- Bent: a non-linear three-atom arrangement, as in water.
- Trigonal planar: three groups around a central atom in one plane, as in boron trifluoride.
- Tetrahedral: four groups arranged around a central atom, as in methane.
- Trigonal pyramidal: three bonded atoms and one lone-pair domain, as in ammonia.
- Trigonal bipyramidal: five groups around a central atom, as in phosphorus pentachloride.
- Octahedral: six groups around a central atom, as in sulfur hexafluoride and many metal complexes.
- Square planar: four groups arranged in a plane, common in some transition-metal complexes.
- Square pyramidal: five-coordinate geometry important in coordination and bioinorganic chemistry.
- T-shaped: a molecular geometry often associated with three bonded atoms and two lone-pair domains in trigonal-bipyramidal electron geometry.
These geometries influence chemical behavior. Tetrahedral carbon supports three-dimensional organic structure. Square-planar metal complexes can show distinctive substitution behavior and stereochemistry. Octahedral complexes are central in coordination chemistry, ligand-field theory, catalysis, and bioinorganic chemistry. Planar conjugated systems support delocalized pi bonding and spectroscopy.
Geometry also affects polarity. A molecule with polar bonds may be nonpolar if its shape causes bond dipoles to cancel. Geometry affects accessibility. A reactive center buried by steric bulk behaves differently from one exposed to solvent or substrate. Geometry affects spectroscopy because vibrational modes, rotational constants, and electronic transitions depend on structure and symmetry.
Molecular geometry is therefore a chemical classification system in its own right. It classifies local structure, not just elemental identity.
Symmetry Operations and Point Groups
Symmetry is the study of transformations that leave an object effectively unchanged. In molecular chemistry, symmetry operations include rotations, reflections, inversions, and improper rotations. A molecule’s symmetry elements and operations can be organized into a point group.
Point groups matter because symmetry constrains molecular behavior. Symmetry helps determine molecular polarity, chirality, vibrational spectra, orbital interactions, selection rules, degeneracy, ligand-field splitting, crystal packing, and reaction pathways. A molecule’s symmetry is not merely a visual property. It is an organizing principle for chemical prediction.
Common symmetry ideas include:
- identity operation: doing nothing to the molecule;
- rotation axis: rotating a molecule by a specified angle so it matches itself;
- mirror plane: reflecting the molecule across a plane;
- center of inversion: inverting all points through a central point;
- improper rotation: combining rotation and reflection.
Molecular point groups are commonly written using Schoenflies notation, such as \(C_{2v}\), \(D_{3h}\), \(T_d\), or \(O_h\). These compact labels summarize symmetry content. Water belongs to \(C_{2v}\). Methane belongs to \(T_d\). Many octahedral complexes are approximated by \(O_h\), though real ligands, distortions, and environments may lower symmetry.
Symmetry can simplify chemical analysis. It can identify whether a molecule has a dipole moment, predict how many infrared or Raman bands may appear, classify molecular orbitals, and help interpret electronic structure. In crystal structures, symmetry expands from point groups to space groups, where translational order is included.
Symmetry is one of the places where chemistry becomes explicitly mathematical. It connects molecular structure to group theory, matrices, spectra, orbitals, degeneracy, and selection rules.
Chirality, Stereochemistry, and Molecular Handedness
Chirality occurs when an object is not superimposable on its mirror image. A chiral molecule has handedness. Many chiral molecules contain a stereocenter, but chirality can also arise from axes, helices, conformations, metal coordination environments, atropisomerism, or larger structural arrangements.
Chirality matters because biological systems are often chiral. Enzymes, receptors, sugars, amino acids, nucleic acids, and many natural products distinguish between mirror-image forms. Two enantiomers of a drug can have different biological effects. In materials and spectroscopy, chirality can affect optical activity, circular dichroism, crystallization, self-assembly, and chiral recognition.
Symmetry and chirality are related. A molecule with an improper rotation axis, mirror plane, or inversion center is generally achiral. Chiral molecules lack certain symmetry elements. Thus, symmetry analysis helps determine whether a structure is chiral.
Stereochemistry also includes cis-trans isomerism, E/Z alkene geometry, R/S configuration, conformational stereochemistry, atropisomerism, axial chirality, helicity, and stereochemical descriptors used in organic, inorganic, biological, and materials chemistry.
Stereochemical identity must be handled carefully in structural communication. A flat drawing may omit wedges, dashes, conformational context, or absolute configuration. A computational structure may represent one stereoisomer while a sample contains a mixture. A crystal may contain one enantiomer, a racemate, or a conglomerate. A protein binding site may select one configuration strongly.
Structure therefore includes not only shape, but handedness and spatial identity.
Molecular Orbitals and Structural Form
Molecular geometry is connected to electronic structure. Atoms arrange themselves in ways that reflect bonding, orbital overlap, electron repulsion, steric effects, and energy minimization. A molecule’s shape is not arbitrary. It is an outcome of electronic interactions and nuclear positions.
Molecular orbital theory describes electrons as occupying orbitals that may extend across an entire molecule. Orbital symmetry affects bonding and reactivity. Overlap between atomic orbitals depends on distance, orientation, energy matching, and symmetry compatibility. Pi systems require geometry that permits overlap of p orbitals. Aromatic systems often require cyclic, planar, conjugated arrangements. Metal-ligand bonding depends on orbital orientation and symmetry.
Structural form can therefore be understood as a compromise among attractive and repulsive interactions. Nuclei repel nuclei. Electrons repel electrons. Electrons attract nuclei. Bonds stabilize. Lone pairs occupy space. Steric bulk creates constraints. Delocalization can favor planarity. Ring strain can distort geometry. Crystal packing can modify molecular conformation.
Orbital thinking also explains why some geometries are reactive. A nucleophile must approach an electrophile along a favorable trajectory. A pericyclic reaction depends on orbital symmetry and geometry. A transition-metal catalyst depends on ligand arrangement and orbital accessibility. A photochemical process may require a geometry that supports excitation and relaxation pathways.
A structural drawing is therefore a simplified representation of a deeper electronic and energetic system. Molecular geometry makes electronic structure spatially interpretable.
Molecular Surfaces and Electron Density
Molecular geometry can be represented through atoms and bonds, but molecules also have surfaces. Space-filling models show approximate occupied volume. Van der Waals surfaces describe steric extent. Solvent-accessible surfaces help represent how molecules interact with solvent or biological environments. Electrostatic potential surfaces show regions of electron richness and deficiency.
Electron density is central because it is closer to the physical distribution of electrons than simple line-bond diagrams. In quantum chemistry, electron density can be used to analyze bonding, charge distribution, polarity, noncovalent interactions, and reactivity. In crystallography, electron-density maps support experimental structure determination.
Different surfaces answer different questions:
- space-filling models emphasize steric volume;
- electrostatic surfaces emphasize charge distribution;
- electron-density maps emphasize evidence and quantum distribution;
- solvent-accessible surfaces emphasize interaction with surrounding media;
- molecular orbital surfaces emphasize electronic states;
- hydrophobic and polar surfaces help interpret binding, solubility, and biomolecular recognition.
Molecular surfaces are especially important in medicinal chemistry, protein structure, catalysis, membrane chemistry, solvent interactions, nanomaterials, and adsorption. A molecule’s surface determines what other molecules “see.” Functional groups, shape complementarity, charge distribution, polar patches, hydrophobic regions, and steric shielding all influence recognition and reactivity.
Molecular structure therefore cannot be reduced to sticks and balls. It also includes fields, densities, boundaries, and interaction surfaces.
Crystal Structure and Extended Order
Many chemical structures are not isolated molecules. Ionic solids, metals, minerals, molecular crystals, coordination polymers, covalent network solids, semiconductors, and metal-organic frameworks require extended structural descriptions.
Crystal structure describes how atoms, ions, or molecules are arranged in repeating three-dimensional order. Important concepts include unit cell, lattice, symmetry, space group, fractional coordinates, packing, coordination environment, intermolecular interactions, and defects. Molecular geometry describes an individual molecule; crystal structure describes how molecular or atomic units are arranged in a solid.
Crystallographic structure is important in pharmaceuticals, materials chemistry, geology, solid-state chemistry, catalysis, energy storage, and semiconductor design. Polymorphs of the same compound can have different solubility, stability, density, melting point, and mechanical properties. In materials, crystal structure can determine conductivity, magnetism, optical behavior, and catalytic activity.
Symmetry appears again in crystallography. Molecules have point groups. Crystals have space groups. Both express the idea that structure is constrained by transformations that preserve order.
Real solids also include disorder, vacancies, surfaces, defects, grain boundaries, strain, hydration, solvation, and thermal motion. A crystallographic structure is a powerful model, but it is still an interpretation of measured diffraction data under assumptions.
For researchers, extended structure matters because many chemical properties are collective. The relevant structure may be a molecule, unit cell, surface, pore, lattice defect, interface, or entire assembly.
Experimental Evidence for Molecular Structure
Molecular structure is supported by evidence. Different methods provide different kinds of structural information.
- X-ray crystallography provides electron-density-based structural information for crystalline materials.
- Neutron diffraction can be especially useful for locating light atoms and studying magnetic or nuclear scattering features.
- Electron diffraction can support structure determination for small crystals or gas-phase systems.
- NMR spectroscopy provides local chemical environments, connectivity, dynamics, conformational information, and distance restraints.
- Infrared and Raman spectroscopy provide vibrational information shaped by molecular geometry and symmetry.
- Microwave spectroscopy can provide precise rotational constants for gas-phase molecules.
- Mass spectrometry supports composition, fragmentation, and structural inference.
- Cryo-electron microscopy supports structural biology and large molecular assemblies.
- Small-angle scattering can support structural information for polymers, colloids, proteins, and soft matter.
- Scanning probe microscopy can provide surface and nanoscale structural information.
No method is universal. Each has assumptions, limitations, sample requirements, and resolution constraints. A high-quality structure is not simply a pretty model; it is an interpreted result from evidence.
Experimental structure determination is also connected to databases. Structural databases allow chemists to compare bond lengths, angles, conformations, crystal packing, coordination geometries, and unusual structures across large bodies of evidence.
For researchers, structural evidence should be interpreted with method awareness. A crystal structure may not equal a solution structure. An NMR-derived ensemble may not show one fixed geometry. A computationally optimized geometry may not reflect thermal motion. An electron-density map requires model fitting and refinement.
Molecular structure is strongest when multiple forms of evidence converge.
Computational Geometry and Structural Modeling
Computational chemistry represents molecular structure using coordinates, graphs, force fields, quantum calculations, conformer searches, geometry optimization, molecular dynamics, docking workflows, structural databases, and data structures. A molecular file may store atom identities, coordinates, bonds, charges, residues, unit cells, symmetry operations, or metadata.
Geometry optimization searches for a structure that minimizes energy under a model. Molecular mechanics uses force fields. Quantum chemistry uses electronic-structure methods. Molecular dynamics simulates motion over time. Conformer generation explores possible low-energy shapes. Docking and molecular recognition workflows evaluate geometric complementarity.
Computational geometry requires caution. A calculated structure is model-dependent. Results may depend on force field, basis set, functional, solvent model, charge state, protonation state, tautomeric form, conformational sampling, convergence criteria, dispersion correction, temperature, pressure, and initial geometry. A structure can look precise while being chemically incomplete.
Reproducible structural modeling requires input files, parameters, software versions, units, assumptions, output validation, and provenance. Molecular geometry is not only visual; it is computationally auditable.
Computational modeling is especially valuable when paired with evidence. A computed structure can help interpret spectra, propose conformations, compare stereoisomers, estimate strain, evaluate reaction pathways, or analyze protein-ligand binding. But it should not be mistaken for experimental truth unless validated against appropriate data.
For researchers, computational structure is a powerful hypothesis engine. Its value depends on transparency, method choice, sampling, validation, and chemical judgment.
Structural Data, Computation, and Reproducibility
Structural chemistry increasingly depends on data systems. Molecular coordinates, crystallographic information files, protein structures, computational outputs, conformer libraries, spectra, structural identifiers, and molecular graphs must be stored, searched, compared, and reused.
Reproducible structural workflows should preserve:
- molecular identity and formula;
- connectivity and bond-order assumptions;
- stereochemistry and tautomer/protonation state;
- atomic coordinates and units;
- coordinate source, method, and uncertainty;
- crystal form, unit cell, space group, and disorder model where relevant;
- computational method, basis set, force field, and software version;
- solvent model, charge state, multiplicity, and boundary conditions;
- conformer generation and filtering criteria;
- geometry optimization convergence status;
- experimental method and instrument metadata;
- validation metrics and provenance.
This matters because structural data can be deceptively portable. A molecule drawn in two dimensions may omit stereochemistry. A coordinate file may lack bond-order information. A protein structure may omit hydrogens. A crystal structure may contain disorder. A computational model may use an incorrect protonation state. A geometry may represent a local minimum rather than the biologically active form.
For researchers, structural data should be treated as evidence with context, not as free-floating coordinates. The most useful structural workflow makes assumptions visible and allows later users to evaluate whether the structure is fit for purpose.
Mathematical Lens: Molecular Geometry, Symmetry, and Structure
Molecular structure can be described through coordinates, vectors, matrices, transformations, and graph relationships. An atomic coordinate is:
\mathbf{r}_i = (x_i, y_i, z_i)
\]
Interpretation: The coordinate vector gives the position of atom \(i\) in three-dimensional space.
A bond distance is:
d_{ij} = \sqrt{(x_i-x_j)^2+(y_i-y_j)^2+(z_i-z_j)^2}
\]
Interpretation: The distance formula calculates separation between atoms \(i\) and \(j\).
A bond angle follows from:
\cos\theta = \frac{\mathbf{u}\cdot\mathbf{v}}{\|\mathbf{u}\|\|\mathbf{v}\|}
\]
Interpretation: The dot product determines the angle between two vectors.
The center of geometry is:
\mathbf{r}_{\mathrm{center}} = \frac{1}{N}\sum_{i=1}^{N}\mathbf{r}_i
\]
Interpretation: The geometric center averages atomic positions without mass weighting.
The center of mass is:
\mathbf{r}_{\mathrm{cm}} = \frac{\sum_i m_i\mathbf{r}_i}{\sum_i m_i}
\]
Interpretation: The center of mass weights each coordinate by atomic mass.
A rotation matrix around the z-axis is:
R_z(\theta)=
\begin{bmatrix}
\cos\theta & -\sin\theta & 0 \\
\sin\theta & \cos\theta & 0 \\
0 & 0 & 1
\end{bmatrix}
\]
Interpretation: Multiplying coordinates by this matrix rotates them by angle \(\theta\) around the z-axis.
A distance matrix is:
D_{ij} = \|\mathbf{r}_i-\mathbf{r}_j\|
\]
Interpretation: A distance matrix stores all pairwise interatomic distances.
An adjacency matrix is:
A_{ij} =
\begin{cases}
1 & \text{if atoms } i \text{ and } j \text{ are bonded}\\
0 & \text{otherwise}
\end{cases}
\]
Interpretation: A molecular graph can represent atoms as nodes and bonds as edges.
Root-mean-square deviation is:
RMSD = \sqrt{\frac{1}{N}\sum_{i=1}^{N}\|\mathbf{r}_i-\mathbf{r}’_i\|^2}
\]
Interpretation: RMSD measures average structural difference between two coordinate sets after appropriate alignment when required.
A transformation of coordinates may be written:
\mathbf{r}’ = R\mathbf{r}+\mathbf{t}
\]
Interpretation: A rotation matrix \(R\) and translation vector \(\mathbf{t}\) can move a structure in space without changing internal geometry.
These equations show how molecular structure becomes computable. A molecular model is not only a picture. It can be represented as coordinates, distances, angles, matrices, transformations, and graph relationships.
Computational Workflows for Molecular Geometry
Computational workflows can make molecular geometry more transparent. A workflow can track atom coordinates, bond distances, bond angles, torsion angles, centers of mass, distance matrices, adjacency matrices, symmetry operations, conformer RMSD, stereochemical labels, structure-source metadata, validation status, and provenance.
Useful workflows include molecular-coordinate parsing, distance matrix calculation, bond-angle calculation, torsion-angle calculation, conformer comparison, RMSD analysis, center-of-mass calculation, rotation-matrix demonstrations, molecular graph construction, stereochemistry checks, crystal-coordinate registries, and SQL evidence systems.
For researchers, structural workflows should preserve four distinctions:
- Formula versus structure: molecular formula does not determine connectivity, stereochemistry, conformation, or phase.
- Drawing versus evidence: a structural diagram is a representation, not the evidence itself.
- Static structure versus ensemble: flexible molecules often require conformational populations, not a single model.
- Experimental structure versus computational structure: both require method context, assumptions, and validation.
The examples below use synthetic educational data. They do not validate real structures, certify stereochemical identity, approve pharmaceutical modeling, establish protein binding, or replace professional structural analysis. They demonstrate how molecular geometry can be organized, audited, and communicated responsibly.
Python Example: Distances, Angles, Torsions, RMSD, and Provenance
The following Python example uses synthetic educational coordinates. It calculates a distance matrix, a water bond angle, a torsion angle for a simplified four-atom chain, RMSD between two conformers, a center of mass, and provenance outputs. In real structural chemistry, these workflows should preserve coordinate sources, units, uncertainty, stereochemistry, protonation state, and validation evidence.
from pathlib import Path
import json
import math
import platform
import sys
import numpy as np
import pandas as pd
# Synthetic molecular geometry workflow.
# Educational example only; not for structural validation,
# pharmaceutical modeling, protein binding claims,
# stereochemical certification, or safety-critical decisions.
def require_columns(data, required, table_name):
"""Raise an error if required columns are missing."""
missing = [column for column in required if column not in data.columns]
if missing:
raise ValueError(f"{table_name} is missing required columns: {missing}")
def distance_matrix(coordinates):
"""Return a pairwise Euclidean distance matrix."""
distances = np.zeros((len(coordinates), len(coordinates)))
for i in range(len(coordinates)):
for j in range(len(coordinates)):
distances[i, j] = np.linalg.norm(coordinates[i] - coordinates[j])
return distances
def angle_degrees(point_a, point_b, point_c):
"""Calculate angle A-B-C in degrees."""
vector_u = point_a - point_b
vector_v = point_c - point_b
cosine = np.dot(vector_u, vector_v) / (
np.linalg.norm(vector_u) * np.linalg.norm(vector_v)
)
cosine = np.clip(cosine, -1.0, 1.0)
return math.degrees(math.acos(cosine))
def torsion_degrees(point_a, point_b, point_c, point_d):
"""Calculate torsion angle A-B-C-D in degrees."""
b1 = point_b - point_a
b2 = point_c - point_b
b3 = point_d - point_c
normal_1 = np.cross(b1, b2)
normal_2 = np.cross(b2, b3)
normal_1 = normal_1 / np.linalg.norm(normal_1)
normal_2 = normal_2 / np.linalg.norm(normal_2)
m1 = np.cross(normal_1, b2 / np.linalg.norm(b2))
x_value = np.dot(normal_1, normal_2)
y_value = np.dot(m1, normal_2)
return math.degrees(math.atan2(y_value, x_value))
def center_of_mass(coordinates, masses):
"""Calculate center of mass."""
return np.sum(coordinates * masses[:, None], axis=0) / np.sum(masses)
def rmsd_no_alignment(coords_a, coords_b):
"""
Calculate RMSD without alignment.
This is transparent for education; production workflows often align structures first.
"""
if coords_a.shape != coords_b.shape:
raise ValueError("Coordinate arrays must have the same shape.")
return float(np.sqrt(np.mean(np.sum((coords_a - coords_b) ** 2, axis=1))))
atoms = pd.DataFrame({
"atom": ["O", "H1", "H2"],
"mass": [15.999, 1.008, 1.008],
"x": [0.000, 0.958, -0.239],
"y": [0.000, 0.000, 0.927],
"z": [0.000, 0.000, 0.000],
})
require_columns(atoms, ["atom", "mass", "x", "y", "z"], "atoms")
coords = atoms[["x", "y", "z"]].to_numpy()
masses = atoms["mass"].to_numpy()
distances = distance_matrix(coords)
distance_table = pd.DataFrame(
distances,
index=atoms["atom"],
columns=atoms["atom"],
)
h_o_h_angle = angle_degrees(coords[1], coords[0], coords[2])
water_center_of_mass = center_of_mass(coords, masses)
four_atom_chain = pd.DataFrame({
"atom": ["A", "B", "C", "D"],
"x": [0.0, 1.5, 2.5, 3.5],
"y": [0.0, 0.0, 1.0, 1.0],
"z": [0.0, 0.0, 0.0, 0.5],
})
chain_coords = four_atom_chain[["x", "y", "z"]].to_numpy()
torsion_angle = torsion_degrees(
chain_coords[0],
chain_coords[1],
chain_coords[2],
chain_coords[3],
)
conformer_a = chain_coords.copy()
conformer_b = np.array([
[0.0, 0.0, 0.0],
[1.5, 0.0, 0.0],
[2.4, 1.1, 0.1],
[3.4, 1.2, 0.7],
])
conformer_rmsd = rmsd_no_alignment(conformer_a, conformer_b)
geometry_summary = pd.DataFrame([{
"structure": "synthetic_water",
"h_o_h_angle_degrees": h_o_h_angle,
"center_of_mass_x": water_center_of_mass[0],
"center_of_mass_y": water_center_of_mass[1],
"center_of_mass_z": water_center_of_mass[2],
}])
torsion_summary = pd.DataFrame([{
"structure": "synthetic_four_atom_chain",
"torsion_A_B_C_D_degrees": torsion_angle,
"conformer_rmsd_no_alignment_angstrom": conformer_rmsd,
}])
output_dir = Path("outputs")
output_dir.mkdir(exist_ok=True)
atoms.to_csv(output_dir / "synthetic_water_coordinates.csv", index=False)
distance_table.to_csv(output_dir / "synthetic_water_distance_matrix.csv")
geometry_summary.to_csv(output_dir / "synthetic_water_geometry_summary.csv", index=False)
four_atom_chain.to_csv(output_dir / "synthetic_chain_coordinates.csv", index=False)
torsion_summary.to_csv(output_dir / "synthetic_torsion_rmsd_summary.csv", index=False)
manifest = {
"workflow": "synthetic_molecular_geometry_workflow",
"data_type": "synthetic educational molecular coordinate records",
"coordinate_unit": "angstrom",
"structures": [
"synthetic_water",
"synthetic_four_atom_chain",
],
"equations": [
"d_ij = norm(r_i - r_j)",
"cos(theta) = dot(u, v)/(norm(u)*norm(v))",
"torsion angle from plane normals",
"center_of_mass = sum(m_i*r_i)/sum(m_i)",
"RMSD = sqrt(mean(sum((r_i - r_i_prime)^2)))",
],
"python_version": sys.version,
"platform": platform.platform(),
"numpy_version": np.__version__,
"pandas_version": pd.__version__,
"output_files": [
"outputs/synthetic_water_coordinates.csv",
"outputs/synthetic_water_distance_matrix.csv",
"outputs/synthetic_water_geometry_summary.csv",
"outputs/synthetic_chain_coordinates.csv",
"outputs/synthetic_torsion_rmsd_summary.csv",
"outputs/molecular_geometry_manifest.json",
],
"responsible_use": [
"Synthetic educational data only.",
"Real structural workflows require validated coordinates, method metadata, uncertainty estimates, stereochemical review, and expert interpretation.",
],
}
with (output_dir / "molecular_geometry_manifest.json").open(
"w",
encoding="utf-8"
) as file:
json.dump(manifest, file, indent=2)
print("Distance matrix in angstroms")
print("----------------------------")
print(distance_table.round(4))
print("\nWater geometry summary")
print("----------------------")
print(geometry_summary.round(6).to_string(index=False))
print("\nTorsion and conformer comparison")
print("--------------------------------")
print(torsion_summary.round(6).to_string(index=False))This workflow demonstrates structural evidence discipline rather than real structure validation. It separates coordinates, distance matrices, bond angles, torsion angles, center of mass, RMSD, and provenance. A real workflow would add coordinate source metadata, structure validation, uncertainty, stereochemical review, conformer alignment, and independent experimental comparison.
R Example: Center of Mass, Rotation, and Conformer Comparison
The following R example uses synthetic educational data to calculate center of mass, apply a rotation matrix, and compare two conformers. In real structural work, these calculations should be tied to validated coordinates, units, alignment conventions, stereochemistry, uncertainty, and method metadata.
# Synthetic molecular geometry scaffold.
# Educational example only; not for structural validation,
# pharmaceutical modeling, protein binding claims,
# stereochemical certification, or safety-critical decisions.
atoms <- data.frame(
atom = c("O", "H1", "H2"),
mass = c(15.999, 1.008, 1.008),
x = c(0.000, 0.958, -0.239),
y = c(0.000, 0.000, 0.927),
z = c(0.000, 0.000, 0.000)
)
coordinate_matrix <- as.matrix(atoms[, c("x", "y", "z")])
center_of_mass <-
colSums(coordinate_matrix * atoms$mass) / sum(atoms$mass)
center_summary <- data.frame(
center_of_mass_x = center_of_mass[["x"]],
center_of_mass_y = center_of_mass[["y"]],
center_of_mass_z = center_of_mass[["z"]]
)
theta <- 120 * pi / 180
rotation_z <- matrix(
c(
cos(theta), -sin(theta), 0,
sin(theta), cos(theta), 0,
0, 0, 1
),
nrow = 3,
byrow = TRUE
)
rotated_coordinates <- coordinate_matrix %*% t(rotation_z)
rotated_atoms <- data.frame(
atom = atoms$atom,
x_rot = rotated_coordinates[, 1],
y_rot = rotated_coordinates[, 2],
z_rot = rotated_coordinates[, 3]
)
conformer_a <- matrix(
c(
0.0, 0.0, 0.0,
1.5, 0.0, 0.0,
2.5, 1.0, 0.0,
3.5, 1.0, 0.5
),
ncol = 3,
byrow = TRUE
)
conformer_b <- matrix(
c(
0.0, 0.0, 0.0,
1.5, 0.0, 0.0,
2.4, 1.1, 0.1,
3.4, 1.2, 0.7
),
ncol = 3,
byrow = TRUE
)
rmsd_no_alignment <- sqrt(mean(rowSums((conformer_a - conformer_b)^2)))
conformer_summary <- data.frame(
comparison = "synthetic_four_atom_conformers",
rmsd_no_alignment_angstrom = rmsd_no_alignment
)
distance_from_com <- sqrt(rowSums(
(
coordinate_matrix -
matrix(center_of_mass, nrow = nrow(atoms), ncol = 3, byrow = TRUE)
)^2
))
atoms$distance_from_center_of_mass <- distance_from_com
dir.create("outputs", showWarnings = FALSE)
write.csv(
atoms,
file = "outputs/r_water_center_of_mass_distances.csv",
row.names = FALSE
)
write.csv(
center_summary,
file = "outputs/r_center_of_mass_summary.csv",
row.names = FALSE
)
write.csv(
rotated_atoms,
file = "outputs/r_rotated_coordinates.csv",
row.names = FALSE
)
write.csv(
conformer_summary,
file = "outputs/r_conformer_rmsd_summary.csv",
row.names = FALSE
)
sink("outputs/r_molecular_geometry_report.txt")
cat("Synthetic Molecular Geometry Scaffold Report\n")
cat("============================================\n\n")
cat("Coordinate records and center-of-mass distances:\n")
print(atoms)
cat("\nCenter of mass summary:\n")
print(center_summary)
cat("\nRotated coordinates:\n")
print(rotated_atoms)
cat("\nConformer comparison:\n")
print(conformer_summary)
cat("\nResponsible-use note:\n")
cat("Synthetic educational data only. Real structural workflows require validated coordinates, method metadata, uncertainty estimates, stereochemical review, and expert interpretation.\n")
sink()
print(atoms)
print(center_summary)
print(rotated_atoms)
print(conformer_summary)This scaffold shows how R can support structural coordinate analysis, rigid transformations, and conformer comparison. The central issue is not the language but the evidence chain. Structural outputs should remain connected to coordinate source, method, stereochemistry, uncertainty, and validation.
SQL Example: Molecular Structure Evidence Register
Molecular geometry becomes more reliable when structures, coordinates, bonds, angles, torsions, conformers, symmetry assignments, stereochemistry, experimental evidence, computational methods, crystal structures, validation records, and interpretation claims are traceable. A simple evidence register can preserve the context needed to audit structural claims.
CREATE TABLE molecular_structure (
structure_id TEXT PRIMARY KEY,
structure_name TEXT NOT NULL,
formula TEXT,
structure_domain TEXT,
structure_type TEXT,
phase_or_context TEXT,
temperature_K REAL,
pressure_bar REAL,
source_uri TEXT,
structure_review_status TEXT,
notes TEXT
);
CREATE TABLE atom_coordinate (
atom_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
atom_label TEXT NOT NULL,
element_symbol TEXT NOT NULL,
x_coordinate REAL,
y_coordinate REAL,
z_coordinate REAL,
coordinate_unit TEXT,
occupancy REAL,
uncertainty_description TEXT,
atom_review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id)
);
CREATE TABLE bond_record (
bond_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
atom_id_1 TEXT NOT NULL,
atom_id_2 TEXT NOT NULL,
bond_order_description TEXT,
bond_length_value REAL,
bond_length_unit TEXT,
bond_review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id),
FOREIGN KEY (atom_id_1) REFERENCES atom_coordinate(atom_id),
FOREIGN KEY (atom_id_2) REFERENCES atom_coordinate(atom_id)
);
CREATE TABLE angle_record (
angle_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
atom_id_1 TEXT NOT NULL,
central_atom_id TEXT NOT NULL,
atom_id_3 TEXT NOT NULL,
angle_degrees REAL,
angle_review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id),
FOREIGN KEY (atom_id_1) REFERENCES atom_coordinate(atom_id),
FOREIGN KEY (central_atom_id) REFERENCES atom_coordinate(atom_id),
FOREIGN KEY (atom_id_3) REFERENCES atom_coordinate(atom_id)
);
CREATE TABLE torsion_record (
torsion_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
atom_id_1 TEXT NOT NULL,
atom_id_2 TEXT NOT NULL,
atom_id_3 TEXT NOT NULL,
atom_id_4 TEXT NOT NULL,
torsion_degrees REAL,
torsion_review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id),
FOREIGN KEY (atom_id_1) REFERENCES atom_coordinate(atom_id),
FOREIGN KEY (atom_id_2) REFERENCES atom_coordinate(atom_id),
FOREIGN KEY (atom_id_3) REFERENCES atom_coordinate(atom_id),
FOREIGN KEY (atom_id_4) REFERENCES atom_coordinate(atom_id)
);
CREATE TABLE conformer_record (
conformer_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
conformer_label TEXT,
relative_energy_kj_mol REAL,
population_fraction REAL,
rmsd_to_reference_angstrom REAL,
conformer_generation_method TEXT,
conformer_review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id)
);
CREATE TABLE symmetry_record (
symmetry_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
point_group TEXT,
symmetry_elements_description TEXT,
chirality_status TEXT,
polarity_status TEXT,
symmetry_assignment_method TEXT,
symmetry_review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id)
);
CREATE TABLE stereochemistry_record (
stereochemistry_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
stereocenter_label TEXT,
stereochemical_descriptor TEXT,
stereochemical_basis TEXT,
stereochemistry_review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id)
);
CREATE TABLE crystal_structure_record (
crystal_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
crystal_system TEXT,
space_group TEXT,
unit_cell_a REAL,
unit_cell_b REAL,
unit_cell_c REAL,
unit_cell_alpha REAL,
unit_cell_beta REAL,
unit_cell_gamma REAL,
polymorph_label TEXT,
disorder_description TEXT,
crystal_review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id)
);
CREATE TABLE structure_evidence_record (
evidence_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
evidence_type TEXT,
method_name TEXT,
instrument_or_software TEXT,
resolution_or_quality_metric TEXT,
dataset_uri TEXT,
uncertainty_description TEXT,
evidence_review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id)
);
CREATE TABLE computational_geometry_record (
computational_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
software_name TEXT,
software_version TEXT,
method_description TEXT,
basis_or_forcefield_description TEXT,
charge_state INTEGER,
spin_multiplicity INTEGER,
solvent_model_description TEXT,
convergence_status TEXT,
input_uri TEXT,
output_uri TEXT,
computational_review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id)
);
CREATE TABLE structural_interpretation_claim (
claim_id TEXT PRIMARY KEY,
structure_id TEXT NOT NULL,
evidence_id TEXT,
computational_id TEXT,
claim_text TEXT,
claim_type TEXT,
confidence_level TEXT,
limitation_notes TEXT,
review_status TEXT,
FOREIGN KEY (structure_id) REFERENCES molecular_structure(structure_id),
FOREIGN KEY (evidence_id) REFERENCES structure_evidence_record(evidence_id),
FOREIGN KEY (computational_id) REFERENCES computational_geometry_record(computational_id)
);
SELECT
s.structure_id,
s.structure_name,
s.formula,
s.structure_type,
s.phase_or_context,
s.temperature_K,
atom.atom_label,
atom.element_symbol,
bond.bond_length_value,
angle.angle_degrees,
torsion.torsion_degrees,
conformer.conformer_label,
conformer.relative_energy_kj_mol,
symmetry.point_group,
symmetry.chirality_status,
stereo.stereochemical_descriptor,
crystal.space_group,
evidence.evidence_type,
evidence.method_name,
comp.method_description,
comp.convergence_status,
claim.claim_type,
claim.confidence_level,
CASE
WHEN s.structure_review_status IS NOT NULL
AND s.structure_review_status != 'pass'
THEN 'structure review required'
WHEN atom.atom_review_status IS NOT NULL
AND atom.atom_review_status != 'pass'
THEN 'atom coordinate review required'
WHEN bond.bond_review_status IS NOT NULL
AND bond.bond_review_status != 'pass'
THEN 'bond review required'
WHEN angle.angle_review_status IS NOT NULL
AND angle.angle_review_status != 'pass'
THEN 'angle review required'
WHEN torsion.torsion_review_status IS NOT NULL
AND torsion.torsion_review_status != 'pass'
THEN 'torsion review required'
WHEN conformer.conformer_review_status IS NOT NULL
AND conformer.conformer_review_status != 'pass'
THEN 'conformer review required'
WHEN symmetry.symmetry_review_status IS NOT NULL
AND symmetry.symmetry_review_status != 'pass'
THEN 'symmetry review required'
WHEN stereo.stereochemistry_review_status IS NOT NULL
AND stereo.stereochemistry_review_status != 'pass'
THEN 'stereochemistry review required'
WHEN crystal.crystal_review_status IS NOT NULL
AND crystal.crystal_review_status != 'pass'
THEN 'crystal structure review required'
WHEN evidence.evidence_review_status IS NOT NULL
AND evidence.evidence_review_status != 'pass'
THEN 'experimental evidence review required'
WHEN comp.convergence_status IS NOT NULL
AND comp.convergence_status != 'pass'
THEN 'computational convergence review required'
WHEN comp.computational_review_status IS NOT NULL
AND comp.computational_review_status != 'pass'
THEN 'computational geometry review required'
WHEN claim.review_status IS NOT NULL
AND claim.review_status != 'reviewed'
THEN 'interpretation review required'
ELSE 'standard review'
END AS molecular_structure_review_status
FROM molecular_structure s
LEFT JOIN atom_coordinate atom
ON s.structure_id = atom.structure_id
LEFT JOIN bond_record bond
ON s.structure_id = bond.structure_id
LEFT JOIN angle_record angle
ON s.structure_id = angle.structure_id
LEFT JOIN torsion_record torsion
ON s.structure_id = torsion.structure_id
LEFT JOIN conformer_record conformer
ON s.structure_id = conformer.structure_id
LEFT JOIN symmetry_record symmetry
ON s.structure_id = symmetry.structure_id
LEFT JOIN stereochemistry_record stereo
ON s.structure_id = stereo.structure_id
LEFT JOIN crystal_structure_record crystal
ON s.structure_id = crystal.structure_id
LEFT JOIN structure_evidence_record evidence
ON s.structure_id = evidence.structure_id
LEFT JOIN computational_geometry_record comp
ON s.structure_id = comp.structure_id
LEFT JOIN structural_interpretation_claim claim
ON s.structure_id = claim.structure_id
ORDER BY molecular_structure_review_status, s.structure_id, atom.atom_label;The purpose of this register is to keep structural interpretation attached to evidence. A molecular structure result should preserve molecular identity, coordinates, bond metrics, angles, torsions, conformers, symmetry, stereochemistry, crystal data, experimental evidence, computational methods, validation status, and interpretation review. Molecular geometry becomes stronger when its evidence trail is structured.
GitHub Repository
The companion repository for this article can support reproducible workflows for coordinate parsing, distance matrices, bond-angle calculations, torsion analysis, center-of-mass calculations, rotation matrices, conformer RMSD, symmetry-operation demonstrations, molecular graph scaffolds, SQL evidence registers, and responsible structural interpretation.
Complete Code Repository
The full code distribution for this article, including selected molecular geometry examples, expanded computational workflows, reproducible data structures, provenance documentation, coordinate calculations, distance and angle metrics, torsion and RMSD scaffolds, SQL evidence registers, and scientific-computing infrastructure, is available on GitHub.
Limits, Uncertainty, and Responsible Interpretation
Molecular geometry is powerful, but it is not self-interpreting. A drawn structure may omit stereochemistry. A Lewis structure may not show geometry. A ball-and-stick model may exaggerate bonds and underrepresent surfaces. A crystallographic structure may reflect solid-state packing rather than solution behavior. A computationally optimized structure may represent a local minimum under a particular model rather than an experimentally validated structure.
Uncertainty enters structural interpretation at many levels: sample purity, phase, temperature, pressure, protonation state, tautomeric form, conformational sampling, crystal disorder, resolution, refinement quality, force-field parameters, quantum method choice, basis set, solvent model, stereochemical assignment, and coordinate precision.
Structure is also conditional. A molecule may have one preferred conformation in a crystal, another in solution, another when protein-bound, and another in the gas phase. A flexible molecule may require an ensemble. A biomolecule may occupy multiple states. A material may contain defects, strain, polymorphs, surfaces, and grain boundaries that do not appear in an idealized model.
Computational structural workflows add additional risks. Coordinates can be compared without alignment. RMSD can be interpreted without considering symmetry. Protonation states can be wrong. Force fields can be inappropriate. Conformer searches can miss important minima. Geometry optimizations can converge to artifacts. Docking poses can look plausible without experimental support.
The computational examples associated with this article are synthetic and educational. They do not validate real structures, certify stereochemical identity, approve pharmaceutical modeling, establish protein binding, or replace professional structural analysis. They are designed to show how molecular geometry can be structured and audited.
Responsible structural interpretation should match claim strength to evidence. A strong structural claim should specify molecular identity, phase, coordinates, method, uncertainty, stereochemistry, conformational assumptions, validation status, and domain of applicability whenever possible.
Conclusion
Molecular geometry, symmetry, and structure explain how atoms become three-dimensional chemical systems. Bond lengths, bond angles, torsion angles, conformations, stereochemistry, symmetry operations, point groups, electron density, molecular surfaces, and crystal packing all shape chemical behavior.
Structure is not simply a drawing. It is a disciplined representation grounded in measurement, computation, and chemical theory. A molecule can be represented through Lewis structures, coordinates, surfaces, graphs, conformers, spectra, crystal structures, and electron densities. Each representation answers a different question.
Modern chemistry is increasingly structural, computational, and data-intensive. Drug discovery depends on three-dimensional complementarity between molecules and biological targets. Materials chemistry depends on crystal structure, porosity, surfaces, defects, and extended order. Catalysis depends on active-site geometry, ligand orientation, transition-state structure, and surface arrangement. Environmental chemistry depends on molecular shape, sorption, degradation, volatility, and speciation.
To understand molecular geometry is to understand chemistry as organized space: atoms arranged into forms that react, recognize, fold, pack, conduct, absorb, catalyze, persist, degrade, and sustain the material world.
Related articles
- What Is Chemistry?
- The Chemical Revolution and the Rise of Modern Chemistry
- Measurement, Quantification, and the Experimental Basis of Chemistry
- Chemical Metrology, Standards, and Reference Materials
- Mathematics for Chemistry and Molecular Systems
- Atoms, Elements, and the Periodic Organization of Matter
- Electronic Structure and the Quantum Foundations of Chemistry
- Chemical Bonding and Molecular Structure
- The Periodic Table and the Logic of Chemical Classification
- Intermolecular Forces and the Chemistry of Condensed Matter
- Stoichiometry and the Quantitative Language of Reactions
- Chemical Thermodynamics and Energetics
- Computational Chemistry and Molecular Modeling
- Analytical Chemistry and the Identification of Matter
Further reading
- Atkins, P., de Paula, J. and Keeler, J. (2018) Atkins’ Physical Chemistry. 11th edn. Oxford: Oxford University Press. Available at: https://global.oup.com/academic/product/atkins-physical-chemistry-9780198769866
- Cotton, F.A. (1990) Chemical Applications of Group Theory. 3rd edn. New York: Wiley. Available at: https://www.wiley.com/en-us/Chemical+Applications+of+Group+Theory%2C+3rd+Edition-p-9780471510949
- Housecroft, C.E. and Sharpe, A.G. (2018) Inorganic Chemistry. 5th edn. Harlow: Pearson. Available at: https://www.pearson.com/en-gb/subject-catalog/p/inorganic-chemistry/P200000003413
- McQuarrie, D.A. and Simon, J.D. (1997) Physical Chemistry: A Molecular Approach. Sausalito: University Science Books. Available at: https://uscibooks.aip.org/books/physical-chemistry-a-molecular-approach/
- Harris, D.C. and Bertolucci, M.D. (1989) Symmetry and Spectroscopy: An Introduction to Vibrational and Electronic Spectroscopy. New York: Dover. Available at: https://store.doverpublications.com/products/9780486661445
- Cambridge Crystallographic Data Centre (n.d.) The Cambridge Structural Database. Available at: https://www.ccdc.cam.ac.uk/solutions/software/csd/
- Protein Data Bank (n.d.) RCSB Protein Data Bank. Available at: https://www.rcsb.org/
- MIT OpenCourseWare (2014) Principles of Chemical Science. Available at: https://ocw.mit.edu/courses/5-111sc-principles-of-chemical-science-fall-2014/
- Royal Society of Chemistry (n.d.) The Shapes of Molecules. Available at: https://edu.rsc.org/ideas/the-shapes-of-molecules/3007452.article
- National Institute of Standards and Technology (n.d.) NIST Chemistry WebBook. Available at: https://webbook.nist.gov/chemistry/
References
- Atkins, P., de Paula, J. and Keeler, J. (2018) Atkins’ Physical Chemistry. 11th edn. Oxford: Oxford University Press. Available at: https://global.oup.com/academic/product/atkins-physical-chemistry-9780198769866
- Cambridge Crystallographic Data Centre (n.d.) The Cambridge Structural Database. Available at: https://www.ccdc.cam.ac.uk/solutions/software/csd/
- Cambridge Crystallographic Data Centre (n.d.) CSD Symmetry. Available at: https://www.ccdc.cam.ac.uk/solutions/software/csdsymmetry/
- Cotton, F.A. (1990) Chemical Applications of Group Theory. 3rd edn. New York: Wiley. Available at: https://www.wiley.com/en-us/Chemical+Applications+of+Group+Theory%2C+3rd+Edition-p-9780471510949
- Harris, D.C. and Bertolucci, M.D. (1989) Symmetry and Spectroscopy: An Introduction to Vibrational and Electronic Spectroscopy. New York: Dover. Available at: https://store.doverpublications.com/products/9780486661445
- Housecroft, C.E. and Sharpe, A.G. (2018) Inorganic Chemistry. 5th edn. Harlow: Pearson. Available at: https://www.pearson.com/en-gb/subject-catalog/p/inorganic-chemistry/P200000003413
- International Union of Pure and Applied Chemistry (n.d.) Compendium of Chemical Terminology: Point Group. Available at: https://goldbook.iupac.org/terms/view/P04703
- International Union of Pure and Applied Chemistry (n.d.) Compendium of Chemical Terminology: Stereochemistry. Available at: https://goldbook.iupac.org/
- International Union of Pure and Applied Chemistry (n.d.) Compendium of Chemical Terminology: Symmetry Number. Available at: https://goldbook.iupac.org/terms/view/S06214
- McQuarrie, D.A. and Simon, J.D. (1997) Physical Chemistry: A Molecular Approach. Sausalito: University Science Books. Available at: https://uscibooks.aip.org/books/physical-chemistry-a-molecular-approach/
- National Center for Biotechnology Information (n.d.) PubChem. Available at: https://pubchem.ncbi.nlm.nih.gov/
- National Institute of Standards and Technology (n.d.) NIST Chemistry WebBook. Available at: https://webbook.nist.gov/chemistry/
- Protein Data Bank (n.d.) RCSB Protein Data Bank. Available at: https://www.rcsb.org/
- Royal Society of Chemistry (2024) Resources for Reasoning of Chemistry Concepts: Multimodal Molecular Geometry. Available at: https://pubs.rsc.org/en/content/articlehtml/2024/rp/d3rp00186e
- Royal Society of Chemistry (n.d.) The Shapes of Molecules. Available at: https://edu.rsc.org/ideas/the-shapes-of-molecules/3007452.article