
Last Updated May 28, 2026
Molecular biology and the flow of genetic information examine how living systems store, transmit, interpret, regulate, repair, and sometimes alter hereditary information across generations and within the life of cells. Molecular biology is one of the central frameworks of modern biology because it explains how biological continuity becomes materially possible and how inherited information becomes cellular function. DNA stores hereditary information in comparatively stable molecular form. RNA mobilizes, processes, regulates, and diversifies that information. Proteins carry out much of the catalytic, structural, transport, signaling, and regulatory work of living systems. But the flow of genetic information is not a rigid one-way script. It is a regulated, conditional, historically contingent, and context-dependent system shaped by replication, transcription, translation, gene regulation, mutation, repair, RNA processing, protein turnover, cellular state, environmental pressure, and evolutionary history.
This article develops Molecular Biology and the Flow of Genetic Information as a foundational article within the Biology knowledge series. It treats the flow of genetic information not merely as the familiar DNA-to-RNA-to-protein sequence, but as one of biology’s major explanatory architectures: the framework through which heredity, development, metabolism, adaptation, disease, biotechnology, ecological response, and systems-level biological organization become experimentally intelligible. The classical central dogma remains essential, but modern molecular biology shows that information flow is regulated, layered, reversible in limited contexts, environmentally responsive, and embedded in larger biological systems.
Main Library
Publications
Article Map
Biology
Related Topic
Chemistry
Related Topic
Earth Science
Related Topic
Environmental Science
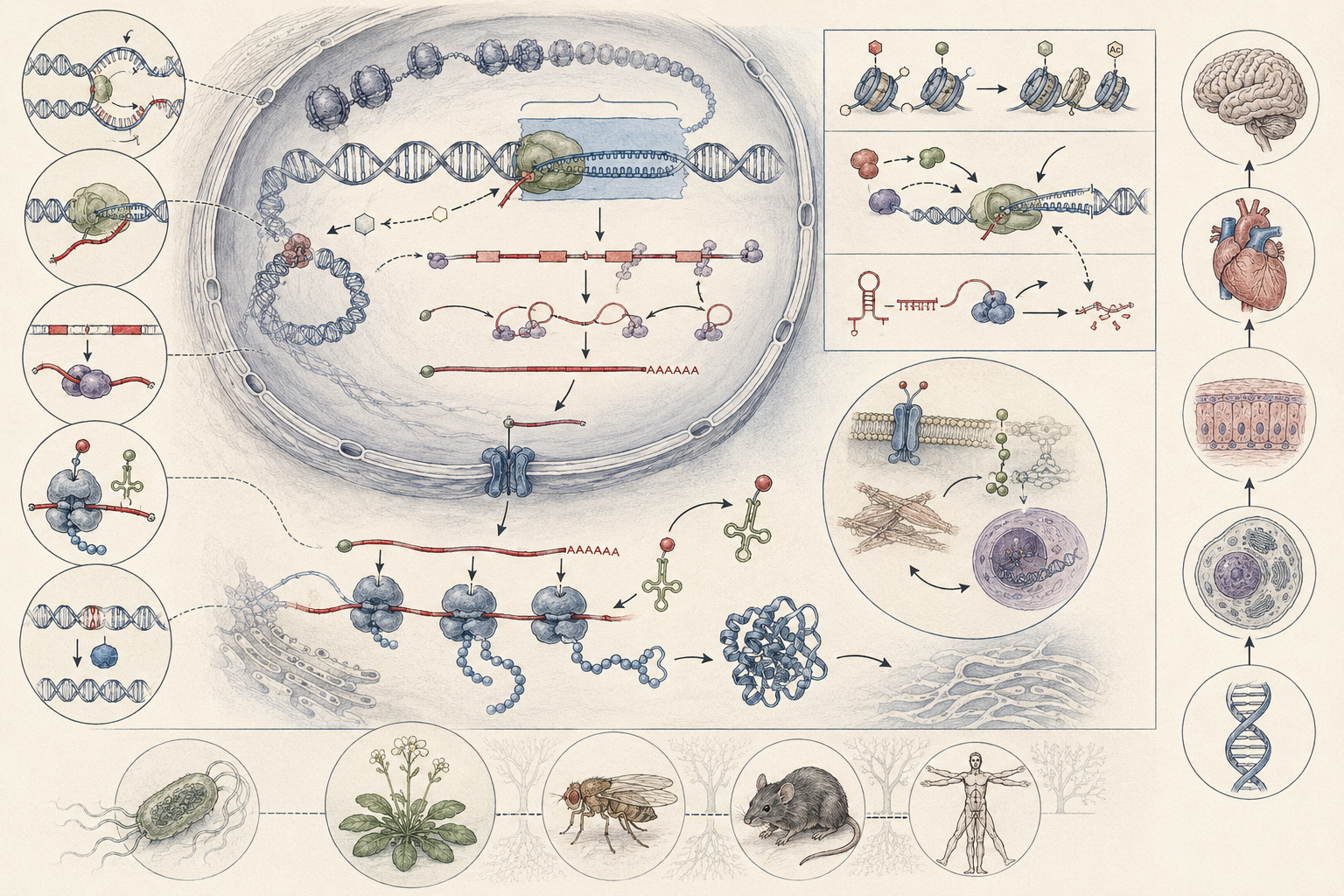
The article develops molecular biology and the flow of genetic information across DNA replication, transcription, RNA processing, translation, the genetic code, ribosomes, gene regulation, chromatin context, mutation, DNA repair, protein synthesis, molecular evolution, cellular differentiation, biotechnology, bioinformatics, molecular diagnostics, ecological molecular response, plant and microbial systems, disease ecology, and computational biology.
The article also extends molecular biology into quantitative and computational analysis through transcript-decay modeling, production-decay expression dynamics, sequence-distance calculation, Jukes-Cantor correction, GC-content analysis, codon-usage summaries, translation scaffolds, mutation-rate reasoning, expression matrices, log2 fold change, PCA-style ordination, information-flow scoring, R workflows, Python workflows, SQL provenance structures, and a linked full-stack GitHub repository containing Python, R, Julia, Fortran, Rust, Go, C, C++, SQL, notebooks, data files, validation notes, and reproducibility documentation.
What Molecular Biology Studies
Molecular biology studies the molecules and molecular systems through which living organisms store hereditary information, regulate cellular activity, build biological structure, and coordinate function across time. At its core, it asks how information is embodied materially in nucleic acids, how that information is copied and transformed, and how molecular processes generate the larger organization of cells, tissues, organisms, populations, and ecosystems. Molecular biology is therefore not simply the study of small things. It is the study of how living systems become biologically specific through molecular organization.
This makes molecular biology one of the central bridges between heredity and function. Genes are not abstract instructions floating outside matter. They exist as physical sequences in DNA, are copied through replication, transcribed into RNA, and often translated into proteins that carry out structural, catalytic, signaling, transport, and regulatory roles. Molecular biology became historically decisive because it made biological inheritance and biological function experimentally tractable at a new level of precision.
Yet molecular biology is not merely about the existence of DNA or the mechanics of protein synthesis. It is about the organized flow of information through living systems: how information is conserved, altered, regulated, repaired, amplified, silenced, processed, translated, degraded, and expressed differently across conditions. In that sense, molecular biology is one of the most powerful ways modern biology understands continuity and change.
Molecular biology also clarifies why biological information is not separate from material conditions. DNA must be replicated by enzymes, RNA must be processed and degraded, ribosomes must translate codons, proteins must fold and function, and repair systems must operate under cellular constraints. Biological information exists only because molecular systems preserve, interpret, and act on it.
DNA, RNA, and Protein as a Framework of Biological Information
The familiar framework of molecular biology is often summarized as the movement of information from DNA to RNA to protein. This formulation remains powerful because it identifies a major organizing principle of cellular life. DNA stores hereditary information in relatively stable form. RNA serves multiple roles in transcriptional output, regulation, catalysis, processing, and translation. Proteins carry out much of the catalytic, structural, transport, signaling, mechanical, and regulatory work of cells. Together, these molecules form a core informational and functional axis of life.
But this framework is best understood as a guiding structure rather than a simplistic one-way chain. RNA is not merely a passive intermediary, and proteins do not simply execute fixed instructions without feedback. Cells regulate transcription, process RNA, modulate translation, alter protein stability, and respond dynamically to internal and external cues. Molecular biology therefore reveals that information flow is real, but it is regulated, conditional, reversible in limited contexts, and system-dependent.
This perspective is important because it prevents two common mistakes. The first is to imagine genes as deterministic scripts that operate independently of context. The second is to imagine molecular biology as so dynamic that stable informational organization disappears altogether. In reality, molecular biology shows both continuity and contingency: stable hereditary storage combined with regulated, context-sensitive expression.
The flow of genetic information is therefore neither mechanical fatalism nor biological chaos. It is an organized system in which molecules preserve, interpret, and modify information under living conditions. The cell is not a machine that merely reads a script; it is a regulated molecular environment in which information is copied, processed, checked, expressed, and sometimes reinterpreted.
Replication and the Continuity of Heredity
Replication is the molecular process by which DNA is copied before cell division, ensuring continuity of hereditary information across cellular generations and, in multicellular organisms, across development and reproduction. It is one of the deepest molecular foundations of biological continuity. Without reliable copying, heredity would be impossible and lineage persistence would collapse into biochemical instability.
Replication matters because it combines fidelity with the possibility of change. The copying process is highly accurate, supported by base pairing, proofreading, and repair systems, yet it is not infinitely perfect. Small errors, if not corrected, become mutations, and these mutations provide part of the raw material for evolution, adaptation, disease, and lineage divergence. Molecular continuity and molecular change therefore arise from the same fundamental process.
This duality is biologically profound. Replication is not merely the preservation of sameness. It is the regulated production of continuity within which variation remains possible. That is why molecular biology links so naturally to both development and evolution. The same process that allows cells and organisms to remain biologically continuous also allows lineages to accumulate change across time.
Replication therefore belongs at the center of molecular biology because it explains how information persists materially while remaining historically open. It also explains why repair, proofreading, chromosomal organization, polymerase fidelity, replication timing, and cell-cycle coordination are not secondary details. They are part of the molecular infrastructure of heredity.
Transcription and the Production of RNA
Transcription is the process by which information in DNA is copied into RNA. It is the first major step by which stored hereditary information becomes functionally available to the cell. Yet transcription is not just copying. It is also selection, timing, and regulation. Cells do not transcribe all genes equally, all the time, or in all conditions. Transcriptional control is one of the major ways biological systems differentiate cell types, respond to stress, regulate metabolism, coordinate development, and adapt to changing environments.
RNA itself is more diverse than a simple messenger concept suggests. Messenger RNA carries coding information for translation, but transfer RNAs, ribosomal RNAs, small RNAs, regulatory RNAs, long noncoding RNAs, and catalytic RNAs all expand the molecular landscape. This means molecular biology is not adequately described as DNA issuing instructions to protein through a single passive intermediate. It is a layered and interactive system in which RNA is central to both expression and regulation.
Transcription therefore reveals one of molecular biology’s key lessons: information in living systems becomes biologically meaningful not simply by existing, but by being selectively mobilized under particular conditions. A gene can be present in every cell of an organism, yet active only in certain tissues, developmental stages, stress states, or environmental contexts.
RNA production is thus one of the first places where the flow of genetic information becomes conditional rather than automatic. The transcriptome is a living snapshot of molecular state: not a full explanation by itself, but a crucial record of what genetic information is being mobilized under specific biological circumstances.
Translation and the Synthesis of Protein
Translation is the process by which nucleotide information in messenger RNA is used to assemble amino acids into proteins. It links informational sequence to molecular function. Through translation, hereditary information becomes enzymes, receptors, channels, structural proteins, transporters, antibodies, transcription factors, signaling proteins, molecular motors, and countless other molecules required for life.
This process is deeply organized. Ribosomes, transfer RNAs, codons, anticodons, initiation factors, elongation machinery, termination signals, aminoacyl-tRNA synthetases, and quality-control systems all contribute to the fidelity and efficiency of protein synthesis. Translation is therefore not simply a chemical conversion. It is a highly coordinated molecular system that turns symbolic sequence into functional material form.
The significance of translation is especially clear because proteins mediate so much of biological activity. When cells grow, divide, repair damage, signal, metabolize nutrients, defend against pathogens, or respond to environments, protein synthesis and protein regulation are usually involved. Translation is thus one of the main points at which hereditary information becomes embodied biological action.
Translation also shows why the flow of genetic information is not merely informational in the abstract. The genetic code becomes life only because molecular machinery interprets it. A codon table printed on a page is not yet biology. Biology requires ribosomes, charged tRNAs, amino acids, energy, quality control, cellular context, and regulated timing.
Gene Regulation and Context-Dependent Expression
Gene regulation is one of the most important themes in modern molecular biology because it explains how the same genome can produce different outcomes in different cells, developmental stages, or environmental conditions. Regulation occurs through many mechanisms, including transcription factors, chromatin state, enhancers, silencers, regulatory RNA, RNA processing, splicing, transcript stability, translational control, protein modification, and controlled degradation. What a gene does therefore depends not only on sequence, but on regulatory context.
This is one reason molecular biology does not support simplistic genetic determinism. Genes matter profoundly, but their effects are mediated through networks of regulation, timing, environment, interaction, and cellular history. A liver cell, a neuron, a root cell, a coral symbiont, and a marine microbe may all rely on DNA and RNA, but they express very different molecular programs because regulation is different.
Regulation is also what allows living systems to remain adaptive rather than chemically rigid. Cells can respond to temperature shifts, nutrient limitation, toxins, pathogens, salinity stress, hypoxia, developmental cues, endocrine signals, or immune activation because molecular expression is conditional and dynamic. In that sense, regulation is one of the major ways life turns stored possibility into context-specific function.
The flow of genetic information is therefore not a pipeline in which every stored sequence becomes protein. It is a regulated interpretive system. Molecular biology is strongest when it studies not only which sequences exist, but when, where, how strongly, and under what conditions those sequences are expressed.
Mutation, Repair, and Molecular Change
Molecular biology must account not only for faithful inheritance, but also for damage, error, and change. DNA is subject to replication errors, chemical modification, radiation damage, recombination events, mobile elements, oxidative stress, and environmental pressure. Without repair systems, informational continuity would deteriorate rapidly. Repair pathways therefore occupy a central place in molecular biology because they preserve viability while also shaping evolutionary possibility.
Mutation is significant because it occupies an intermediate space between disorder and novelty. At one level, mutation can disrupt function, produce disease, impair development, alter regulation, or destabilize genomes. At another level, mutation contributes to adaptation, diversity, and long-term evolutionary change. Molecular biology thus reveals that change at the sequence level can be both pathological and generative, depending on context, scale, and consequence.
Repair is equally important because heredity depends not only on copying but on maintenance. DNA repair, proofreading, recombination repair, damage-response signaling, and genome surveillance all protect informational continuity. But repair systems are themselves biological systems with limits, tradeoffs, and evolutionary histories.
This tension between fidelity and change is one of the deepest features of living systems. Molecular biology does not merely explain how organisms remain the same. It also explains how they become different. The flow of genetic information is therefore historical as well as cellular: it preserves lineage, generates variation, and records the consequences of environmental and evolutionary pressure.
Molecular Biology in Physiology, Development, and Biological Function
Molecular biology is not an isolated layer beneath “real” biology. It is one of the major ways physiology, development, and function become scientifically intelligible. Hormone responses depend on receptors and transcriptional programs. Development depends on regulated gene expression across space and time. Metabolism depends on enzymes whose synthesis and control are molecularly mediated. Immunity depends on receptor diversity, signaling cascades, and gene activation. Neural function depends on molecular transport, membrane proteins, RNA localization, protein turnover, and regulated synaptic machinery.
Development makes this especially clear. The emergence of tissues, organs, and body plans depends on differential gene expression, positional information, chromatin state, signaling, RNA regulation, and regulatory coordination. A multicellular organism does not arise because every cell does the same thing. It arises because similar genomes are interpreted differently across developmental contexts.
Molecular biology therefore does not reduce organisms to sequence alone. It helps explain how sequence, regulation, and interaction generate higher-order biological form and function. A molecular explanation becomes biologically strong when it connects information flow to cell behavior, tissue organization, physiological performance, and environmental response.
This is why molecular biology connects naturally to systems biology. The flow of genetic information is not merely a set of molecular events. It is part of a larger biological system in which genes, transcripts, proteins, metabolites, signals, cells, organisms, and environments interact.
Ecology, Evolution, and Sustainability-Adjacent Biology
Molecular biology is deeply relevant to ecology and sustainability-adjacent biology because genes and regulatory systems are part of how organisms respond to environments, adapt to disturbance, tolerate stress, and persist across changing conditions. Ecological resilience, adaptation to climate stress, resistance to pathogens, nutrient-use efficiency, microbial decomposition, host-symbiont interaction, pollutant response, and reproductive timing all have molecular dimensions.
Evolutionary biology is especially important here because molecular change is one of the main substrates of long-term adaptation. Sequence variation, gene duplication, regulatory change, horizontal gene transfer, mobile elements, and selection on expression patterns all contribute to the diversification of life. Molecular biology thus gives ecological and evolutionary thinking a mechanistic layer without replacing population- or ecosystem-level explanation.
This matters for sustainability because living systems do not respond to environmental disruption only at visible organismal scales. They also respond molecularly through stress pathways, altered regulation, repair systems, epigenetic shifts, and selection on variants. A sustainability-oriented biology therefore benefits from seeing molecular biology not as detached from environmental life, but as one of the scales at which environmental constraint becomes biologically real.
This is especially important under rapid environmental change. Warming, acidification, drought, pollution, hypoxia, salinization, habitat fragmentation, invasive species, and pathogen shifts all produce molecular responses before they become visible as population decline, disease emergence, or ecosystem reorganization.
Marine, Freshwater, Soil, Plant, and Microbial Relevance
Marine biology makes the ecological significance of molecular biology especially visible. Marine microbes, phytoplankton, coral symbionts, fishes, invertebrates, and aquatic plants all regulate gene expression in response to temperature, salinity, nutrient limitation, oxygen stress, acidification, toxins, symbiosis, and pathogen exposure. Marine systems therefore reveal that molecular biology is not merely laboratory biology. It is also part of how ocean life persists, adapts, and sometimes fails under environmental pressure.
Freshwater biology shows similar dynamics in rivers, lakes, wetlands, and sediments, where microbial communities, aquatic plants, algae, and animals respond molecularly to eutrophication, pollution, hypoxia, hydrologic change, invasive species pressure, and ecological disturbance. Soil biology is equally molecular, since decomposition, nutrient cycling, plant-microbe interaction, carbon stabilization, fungal exchange, and microbial resilience all depend on gene-regulated metabolic pathways.
Plant science and agroecology also depend strongly on molecular biology. Photosynthesis, stress tolerance, nutrient uptake, root-microbe signaling, flowering, growth regulation, disease resistance, and seed development all involve tightly regulated flows of genetic information. Forestry, restoration ecology, conservation biology, and food systems therefore increasingly rely on molecular methods and molecular understanding.
Across these domains, molecular biology helps explain how environmental pressure becomes biochemical response, gene regulation, molecular damage, physiological adjustment, and evolutionary possibility. It also helps explain why biodiversity cannot be reduced to visible forms alone. Hidden molecular variation, regulatory capacity, microbial function, and stress-response potential all shape living resilience.
Medical, Biomedical, and Disease Ecology Relevance
Molecular biology is foundational to medicine and biomedicine because disease, development, immunity, infection, and therapy all depend on molecular mechanisms. Cancer involves mutation, dysregulated signaling, altered expression, epigenetic change, and genomic instability. Infectious disease involves host-pathogen interaction at molecular interfaces. Immunity depends on recognition systems, gene rearrangement, signaling cascades, and regulated effector responses. Pharmacology increasingly depends on molecular targets and pathway-specific intervention.
Molecular biology also reshaped diagnostics by enabling sequencing, amplification, expression profiling, biomarker analysis, and pathogen detection at high resolution. Clinical reasoning is often still organismal and physiological, but the explanatory depth behind modern diagnosis and treatment is frequently molecular.
Disease ecology provides a broader frame. Pathogens evolve through molecular change, hosts respond through regulated defense, and environmental conditions shape transmission, selection, and persistence. Molecular biology therefore helps connect cellular and clinical thinking to population and ecological dynamics.
This is one reason molecular biology matters far beyond the laboratory. It is part of how medicine, public health, epidemiology, conservation, and ecological resilience interpret biological risk. It helps scientists distinguish exposure from infection, infection from immune response, genetic predisposition from expression state, and molecular mechanism from clinical outcome.
Biotechnology, Bioinformatics, and Computational Relevance
Biotechnology extends molecular biology into applied systems of analysis, intervention, and design. Sequencing, PCR, cloning, gene editing, synthetic pathway assembly, fermentation optimization, molecular diagnostics, metagenomics, biosensor development, protein engineering, and RNA-based technologies all depend on understanding how genetic information is stored, processed, regulated, and manipulated. In biotechnology, molecules of heredity become not only objects of explanation, but tools of engineering and measurement.
Bioinformatics strengthens this transformation by allowing sequence comparison, alignment, assembly, annotation, expression analysis, variant calling, phylogenetic inference, codon analysis, transcript quantification, regulatory-network analysis, and systems modeling at scales impossible without computation. DNA, RNA, and protein are therefore not only molecules in cells. They are also high-density forms of biological data.
This computational turn is especially important because it changes what biological evidence can be. A sequence database, transcriptomic matrix, codon-usage profile, variant table, or regulatory network can now be analyzed alongside microscopy, physiology, ecology, and field observation. Molecular biology thus becomes one of the central meeting points of life science and data science.
This also raises the importance of provenance. Sequence data, expression matrices, variant calls, and transcriptomic summaries require metadata about sample source, extraction method, sequencing platform, preprocessing, normalization, reference genome, filtering criteria, and analytical assumptions. Without transparent workflows, molecular evidence becomes difficult to reproduce or interpret.
Mathematical Lens
Modern molecular biology is not only mechanistic. It is also quantitative. Replication rates, transcriptional output, translation efficiency, mutation frequency, gene-expression change, RNA half-life, protein turnover, codon usage, GC content, and sequence similarity can all be analyzed mathematically and computationally. This does not reduce biology to numbers, but it does make molecular processes more explicit, testable, reproducible, and comparable.
One simple model for transcript decay is exponential loss:
Interpretation: Transcript abundance declines through time according to initial abundance and decay rate.
where \(m(t)\) is transcript abundance at time \(t\), \(m_0\) is initial abundance, and \(k\) is the decay constant. The corresponding half-life is:
Interpretation: Transcript half-life is the time required for RNA abundance to decline by half.
This kind of model is useful because RNA abundance reflects not only production but also degradation. Molecular biology often requires both.
A simple production-decay model for transcript abundance is:
Interpretation: Transcript abundance reflects a balance between time-dependent production and degradation.
where \(\alpha(t)\) is the time-dependent production rate and \(\beta\) is the degradation constant. This is useful because gene expression often reflects a dynamic balance between synthesis, processing, export, translation, and decay rather than one static abundance value.
A common expression-change measure is:
Interpretation: Log2 fold change converts expression ratios into a symmetric scale for comparing conditions.
where \(E_1\) and \(E_2\) are expression values in two conditions and \(\epsilon\) is a small pseudocount.
A simple observed sequence difference between equal-length sequences can be written as:
Interpretation: Observed sequence difference is the fraction of aligned positions that differ.
where \(m\) is the number of mismatches and \(L\) is sequence length. A Jukes-Cantor correction gives:
Interpretation: Jukes-Cantor correction adjusts observed difference for hidden substitutions under a simple evolutionary model.
This is useful because raw mismatch counts can underestimate deeper divergence when multiple substitutions occur at the same site over time.
A simple DNA GC-content measure is:
Interpretation: GC content measures the fraction of DNA bases that are guanine or cytosine.
For RNA, \(U\) replaces \(T\). This is useful because nucleotide composition can influence thermodynamics, codon usage, sequencing behavior, genome organization, and comparative molecular interpretation.
For a codon \(c\), a simple codon frequency is:
Interpretation: Codon frequency measures how often a codon appears relative to all codons in a sequence or dataset.
where \(n_c\) is the count of codon \(c\), and \(\sum_j n_j\) is the total number of codons observed. This is useful because coding sequences carry compositional and translational patterns as well as amino-acid information.
A simple mutation-rate estimate can be written as:
Interpretation: Mutation rate estimates observed mutations relative to genomes, sites, and generations surveyed.
where \(m\) is the number of observed mutations, \(N\) is the number of genomes or individuals observed, \(L\) is the number of sites surveyed, and \(g\) is the number of generations or replication cycles. This is useful because molecular change must often be interpreted relative to opportunity for mutation.
Variables, Units, and Molecular Interpretation
Quantitative molecular biology depends on variables that connect abundance, decay, sequence composition, expression change, mutation, and evolutionary comparison to biological interpretation. The table below summarizes several central quantities.
| Symbol or Term | Meaning | Typical Unit or Scale | Molecular Interpretation |
|---|---|---|---|
| \(m(t)\) | Transcript abundance at time \(t\) | counts, TPM, normalized expression, fluorescence, or arbitrary units | Amount of RNA transcript remaining or measured at a given time |
| \(m_0\) | Initial transcript abundance | same as \(m(t)\) | Starting abundance before decay or perturbation |
| \(k\) | Decay constant | per unit time | Rate of exponential transcript loss |
| \(t_{1/2}\) | Half-life | time | Time required for transcript abundance to decline by half |
| \(\alpha(t)\) | Production rate | abundance per time | Time-dependent rate of transcript synthesis or production |
| \(\beta\) | Degradation constant | per unit time | Rate of transcript removal in production-decay models |
| \(E_1, E_2\) | Expression values in two conditions | counts, normalized counts, TPM, FPKM, or assay units | Molecular expression levels being compared across conditions |
| \(\epsilon\) | Pseudocount | same as expression value | Small value used to avoid division by zero in fold-change calculations |
| \(\log_2 FC\) | Log2 fold change | log2 ratio | Symmetric expression-change measure across conditions |
| \(d\) | Observed sequence distance | fraction from 0 to 1 | Share of aligned positions with mismatches |
| \(d_{\mathrm{JC}}\) | Jukes-Cantor corrected distance | substitutions per site under a simple model | Corrected molecular distance accounting for hidden substitutions |
| \(G,C,A,T,U\) | Nucleotide counts | counts | Observed number of nucleotide bases in DNA or RNA sequence |
| \(GC\) | GC content | fraction or percentage | Share of DNA or RNA sequence composed of guanine and cytosine |
| \(f_c\) | Codon frequency | fraction or percentage | Relative use of codon \(c\) in a coding sequence or dataset |
| \(\mu\) | Mutation rate | mutations per site per generation or replication cycle | Mutation frequency normalized by opportunity for mutation |
| \(N\) | Number of genomes or individuals observed | count | Sample size for mutation-rate estimation |
| \(L\) | Sequence length or surveyed sites | base pairs, nucleotides, amino acids, or sites | Number of positions available for comparison or mutation |
| \(g\) | Generations or replication cycles | count | Number of opportunities for mutations across time |
The table shows why molecular data require careful interpretation. A transcript count, distance value, mutation-rate estimate, or fold-change score becomes biologically meaningful only when linked to sampling design, normalization, organism, tissue, environment, and measurement method.
Worked Example: Transcript Half-Life and Sequence Distance
Suppose a transcript starts at \(m_0=100\) arbitrary units and declines to \(25\) units after 4 hours. Under the exponential decay model:
Interpretation: Transcript abundance declines exponentially when decay rate is approximately constant.
Substituting the values:
Interpretation: The observed decline can be used to estimate the transcript decay constant.
Dividing both sides by 100:
Interpretation: The transcript has declined to 25 percent of its initial abundance.
Taking the natural logarithm:
Interpretation: Log transformation converts the exponential decay relation into a solvable linear expression.
Solving:
Interpretation: The estimated decay constant is approximately 0.3466 per hour.
The half-life is:
Interpretation: The transcript half-life is approximately 2 hours under these conditions.
This is useful because it turns expression decline into an interpretable dynamic parameter rather than a descriptive observation alone.
Sequence comparison can be analyzed similarly. Suppose two equal-length DNA sequences have 3 mismatches across 30 aligned sites. Then:
Interpretation: The observed p-distance is 0.10, meaning 10 percent of aligned sites differ.
A Jukes-Cantor correction gives:
Interpretation: The corrected distance is slightly larger because it accounts for possible hidden substitutions.
This is useful because molecular comparison often begins with observed difference but must consider hidden substitutions over evolutionary time.
Computational Modeling
Computational modeling helps make molecular biology explicit because genetic information can be copied, counted, compared, translated, summarized, normalized, and analyzed across many scales. Transcript-decay models estimate RNA stability. Fold-change calculations compare expression across conditions. PCA-style ordination summarizes expression profiles. Sequence-distance calculations compare molecular similarity. Codon-usage summaries connect nucleotide sequence to translation patterns. Mutation-rate formulas relate observed molecular change to opportunity for mutation.
The selected examples below focus on compact, reusable workflows: transcript half-life estimation, expression matrix summarization, log2 fold change, PCA-style ordination, codon usage, GC content, sequence distance, Jukes-Cantor correction, translation scaffolds, and mutation-rate-style reasoning. The GitHub repository extends the same logic into richer workflows for production-decay expression dynamics, sequence-distance matrices, transcriptomics metadata, SQL provenance, notebooks, validation scripts, and multi-language scientific-computing examples.
The purpose is not to reduce molecular biology to code. The purpose is to make molecular evidence inspectable. A molecular claim becomes stronger when sequences, counts, normalization methods, model assumptions, sample metadata, and analytical code are documented together.
R Workflow: Transcript Decay, Expression Change, PCA Scaffold, and Codon Usage
R is useful for molecular biology because it supports statistical modeling, expression summaries, matrix workflows, reproducible reporting, and sequence-feature calculations. The following workflow estimates transcript half-life, compares expression across conditions, creates a PCA-style ordination scaffold, and calculates codon usage and GC content.
# Molecular Biology and the Flow of Genetic Information Workflow
#
# This workflow demonstrates four quantitative molecular-biology tasks:
#
# 1. Fit an exponential transcript-decay model and estimate half-life.
# 2. Summarize an expression matrix and calculate log2 fold change.
# 3. Create a PCA-style ordination scaffold from expression data.
# 4. Calculate codon usage and GC content from a coding sequence.
#
# These examples can be adapted for transcriptomics, molecular ecology,
# microbial gene-expression studies, plant stress biology, biotechnology,
# diagnostic assay interpretation, and computational biology.
library(tibble)
library(dplyr)
library(tidyr)
library(stringr)
# ------------------------------------------------------------
# 1. Transcript decay and half-life estimation
# ------------------------------------------------------------
decay_df <- tibble(
time_h = c(0, 1, 2, 3, 4),
expr = c(100, 70, 50, 35, 25)
)
decay_fit <- lm(log(expr) ~ time_h, data = decay_df)
k_est <- -coef(decay_fit)[["time_h"]]
m0_est <- exp(coef(decay_fit)[["(Intercept)"]])
half_life_h <- log(2) / k_est
decay_summary <- tibble(
k_est = k_est,
m0_est = m0_est,
half_life_h = half_life_h,
r_squared_log_space = summary(decay_fit)$r.squared
)
decay_result <- decay_df %>%
mutate(
predicted_expr = exp(predict(decay_fit)),
residual = expr - predicted_expr
)
# ------------------------------------------------------------
# 2. Expression matrix, log2 fold change, and PCA scaffold
# ------------------------------------------------------------
set.seed(42)
genes <- paste0("gene_", 1:150)
samples <- paste0("sample_", 1:8)
group <- c(rep("control", 4), rep("treated", 4))
expr_mat <- matrix(
rpois(150 * 8, lambda = 90),
nrow = 150,
ncol = 8,
dimnames = list(genes, samples)
)
# Add synthetic treatment-responsive structure.
expr_mat[1:12, 5:8] <- expr_mat[1:12, 5:8] + 45
expr_mat[13:24, 5:8] <- pmax(expr_mat[13:24, 5:8] - 25, 1)
expr_df <- as.data.frame(expr_mat) %>%
tibble::rownames_to_column("gene")
meta_df <- tibble(sample = samples, group = group)
long_expr_df <- expr_df %>%
pivot_longer(-gene, names_to = "sample", values_to = "count") %>%
left_join(meta_df, by = "sample")
gene_summary_df <- long_expr_df %>%
group_by(gene, group) %>%
summarise(mean_count = mean(count), .groups = "drop") %>%
pivot_wider(names_from = group, values_from = mean_count) %>%
mutate(
log2_fc = log2((treated + 1) / (control + 1)),
mean_expression = (treated + control) / 2
) %>%
arrange(desc(abs(log2_fc)))
log_expr <- log2(expr_mat + 1)
pca <- prcomp(t(log_expr), center = TRUE, scale. = TRUE)
pca_df <- as.data.frame(pca$x[, 1:2]) %>%
tibble::rownames_to_column("sample") %>%
left_join(meta_df, by = "sample")
# ------------------------------------------------------------
# 3. Codon usage and GC content
# ------------------------------------------------------------
coding_seq <- "ATGGCCGCCGAACTGATCGTCAAGGGTAAACCCGGGTTTAA"
codons <- str_sub(
coding_seq,
seq(1, nchar(coding_seq), by = 3),
seq(3, nchar(coding_seq), by = 3)
)
codons <- codons[codons != ""]
codon_df <- tibble(codon = codons) %>%
count(codon, sort = TRUE) %>%
mutate(fraction = n / sum(n))
bases <- str_split(coding_seq, "", simplify = TRUE)
gc_summary <- tibble(
sequence_length = nchar(coding_seq),
gc_fraction = mean(bases %in% c("G", "C"))
)
print(round(decay_summary, 4))
print(round(decay_result, 4))
print(gene_summary_df %>% slice_head(n = 20))
print(pca_df)
print(codon_df)
print(gc_summary)This R workflow is useful because molecular biology increasingly studies expression programs rather than one transcript at a time. It also keeps sequence-level features connected to reproducible data summaries.
Python Workflow: Sequence Distance, Transcript Half-Life, Expression Matrix, and Translation
Python is useful for molecular biology because it supports sequence parsing, numerical modeling, matrix operations, pipeline design, and reproducible computational analysis. The following workflow calculates sequence distance and Jukes-Cantor correction, estimates transcript half-life and expression area under the curve, summarizes an expression matrix, and translates a short coding sequence while reporting codon usage and GC content.
"""
Molecular Biology and the Flow of Genetic Information Workflow
This workflow demonstrates four quantitative molecular-biology tasks:
1. Calculate observed sequence distance and Jukes-Cantor correction.
2. Estimate transcript half-life and expression area under the curve.
3. Summarize an expression matrix with log2 fold change and PCA-style ordination.
4. Translate a coding sequence and summarize codon usage and GC content.
The examples are compact, but the same structures can be extended to
transcriptomics, molecular ecology, microbial gene-expression studies,
plant stress biology, diagnostics, biotechnology, and computational biology.
"""
from __future__ import annotations
from collections import Counter
import numpy as np
import pandas as pd
def sequence_distance_example() -> pd.DataFrame:
"""
Calculate Hamming distance, observed p-distance, and Jukes-Cantor distance.
"""
seq1 = "ATGCTAGCTAACGGTACCTA"
seq2 = "ATGCTGGCTATCGGTACCTA"
if len(seq1) != len(seq2):
raise ValueError("Sequences must be equal length for this simple example.")
mismatches = sum(base_a != base_b for base_a, base_b in zip(seq1, seq2))
length = len(seq1)
p_distance = mismatches / length
if p_distance >= 0.75:
jukes_cantor = np.nan
else:
jukes_cantor = -(3.0 / 4.0) * np.log(1.0 - (4.0 / 3.0) * p_distance)
return pd.DataFrame(
{
"sequence_1": [seq1],
"sequence_2": [seq2],
"length": [length],
"mismatches": [mismatches],
"p_distance": [p_distance],
"jukes_cantor_distance": [jukes_cantor],
}
)
def transcript_half_life_example() -> tuple[pd.DataFrame, pd.DataFrame]:
"""
Estimate transcript half-life and expression area under the curve.
"""
time_h = np.array([0, 1, 2, 3, 4], dtype=float)
expr = np.array([100, 70, 50, 35, 25], dtype=float)
slope, intercept = np.polyfit(time_h, np.log(expr), 1)
k_est = -slope
m0_est = np.exp(intercept)
half_life_h = np.log(2.0) / k_est
auc = np.trapz(expr, time_h)
predicted = np.exp(intercept + slope * time_h)
summary = pd.DataFrame(
{
"k_est": [k_est],
"m0_est": [m0_est],
"half_life_h": [half_life_h],
"AUC": [auc],
}
)
trace = pd.DataFrame(
{
"time_h": time_h,
"expr_observed": expr,
"expr_predicted": predicted,
"residual": expr - predicted,
}
)
return summary, trace
def expression_matrix_example() -> tuple[pd.DataFrame, pd.DataFrame]:
"""
Create an expression matrix, summarize log2 fold change, and build PCA-style scores.
"""
rng = np.random.default_rng(7)
n_genes = 180
n_samples = 10
groups = np.array(["control"] * 5 + ["treated"] * 5)
expr = rng.poisson(lam=90, size=(n_genes, n_samples)).astype(float)
# Add synthetic treatment-responsive structure.
expr[:15, 5:] += 40
expr[15:30, 5:] = np.maximum(expr[15:30, 5:] - 25, 1)
sample_names = [f"sample_{i + 1}" for i in range(n_samples)]
gene_names = [f"gene_{i + 1}" for i in range(n_genes)]
expr_df = pd.DataFrame(expr, index=gene_names, columns=sample_names)
control_mean = expr_df.iloc[:, :5].mean(axis=1)
treated_mean = expr_df.iloc[:, 5:].mean(axis=1)
gene_summary = pd.DataFrame(
{
"gene": gene_names,
"control_mean": control_mean.values,
"treated_mean": treated_mean.values,
"log2_fc": np.log2((treated_mean.values + 1.0) / (control_mean.values + 1.0)),
}
).sort_values("log2_fc", ascending=False)
log_expr = np.log2(expr_df + 1.0)
# PCA-style ordination using SVD.
x = log_expr.sub(log_expr.mean(axis=1), axis=0).T.values
x_centered = x - x.mean(axis=0, keepdims=True)
u, s, _ = np.linalg.svd(x_centered, full_matrices=False)
scores = u[:, :2] * s[:2]
pca_df = pd.DataFrame(
{
"sample": sample_names,
"group": groups,
"PC1": scores[:, 0],
"PC2": scores[:, 1],
}
)
return gene_summary, pca_df
def translation_and_codon_usage_example() -> tuple[pd.DataFrame, pd.DataFrame]:
"""
Translate a short coding sequence and summarize codon usage and GC content.
"""
coding_seq = "ATGGCCGCCGAACTGATCGTCAAGGGTAAACCCGGGTTTAA"
codon_table = {
"ATG": "M",
"GCC": "A",
"GAA": "E",
"CTG": "L",
"ATC": "I",
"GTC": "V",
"AAG": "K",
"GGT": "G",
"AAA": "K",
"CCC": "P",
"GGG": "G",
"TTT": "F",
"TAA": "*",
}
codons = [
coding_seq[i : i + 3]
for i in range(0, len(coding_seq) - 2, 3)
]
protein = "".join(codon_table.get(codon, "X") for codon in codons)
codon_counts = Counter(codons)
codon_df = pd.DataFrame(
{
"codon": list(codon_counts.keys()),
"count": list(codon_counts.values()),
}
).sort_values("count", ascending=False)
codon_df["fraction"] = codon_df["count"] / codon_df["count"].sum()
gc_fraction = sum(base in {"G", "C"} for base in coding_seq) / len(coding_seq)
sequence_summary = pd.DataFrame(
{
"coding_sequence": [coding_seq],
"protein": [protein],
"sequence_length_nt": [len(coding_seq)],
"codon_count": [len(codons)],
"gc_fraction": [gc_fraction],
}
)
return sequence_summary, codon_df
def main() -> None:
"""
Run compact molecular-biology workflows.
"""
sequence_distance = sequence_distance_example()
decay_summary, decay_trace = transcript_half_life_example()
gene_summary, pca_df = expression_matrix_example()
sequence_summary, codon_df = translation_and_codon_usage_example()
print("Sequence distance:")
print(sequence_distance.round(4).to_string(index=False))
print("\nTranscript half-life summary:")
print(decay_summary.round(4).to_string(index=False))
print(decay_trace.round(4).to_string(index=False))
print("\nTop expression changes:")
print(gene_summary.head(20).round(4).to_string(index=False))
print("\nPCA-style ordination:")
print(pca_df.round(4).to_string(index=False))
print("\nTranslation and sequence summary:")
print(sequence_summary.round(4).to_string(index=False))
print("\nCodon usage:")
print(codon_df.round(4).to_string(index=False))
if __name__ == "__main__":
main()This Python workflow is useful because it connects molecular sequence, expression dynamics, condition comparison, and translation in one reproducible scaffold. The examples are compact, but the same logic can scale to transcriptomics, genomics, molecular ecology, diagnostics, and bioinformatics pipelines.
GitHub Repository
The article body includes compact R and Python examples so the biological and scientific argument remains readable. The full repository expands those examples into a broader computational molecular-biology workflow, including transcript-decay fitting, production-decay expression dynamics, half-life estimation, expression matrices, log2 fold change, PCA-style ordination, sequence-distance matrices, Jukes-Cantor correction, GC-content calculation, codon-usage summaries, DNA-to-protein translation scaffolds, mutation-rate examples, molecular-information-flow scoring, SQL provenance structures, validation notes, reproducible data files, and full-stack scientific-computing examples across Python, R, Julia, Fortran, Rust, Go, C, C++, SQL, and notebooks.
Complete Code Repository
The full code distribution for this article, including selected article examples, expanded computational workflows, reproducible data structures, provenance documentation, validation notes, and full-stack scientific-computing scaffolding, is available on GitHub.
Limits, Complexity, and Modern Molecular Thinking
Molecular biology is foundational, but it is not sufficient by itself for all biological explanation. Sequence does not explain everything without regulatory context. Expression data do not automatically explain physiology. Molecular association does not always imply organismal causation. A transcriptomic shift may or may not produce an ecological outcome, and a variant of statistical significance may or may not be biologically decisive.
This is why modern molecular thinking increasingly emphasizes integration rather than reduction. Molecular biology is strongest when it is linked to cell biology, physiology, development, ecology, evolution, environmental context, and systems biology. The point is not to deny molecular importance, but to place molecular information within the larger systems that make biological meaning possible.
Models and workflows are useful because they clarify assumptions, expose patterns, and make comparison possible. But a transcript-decay estimate is not a full theory of regulation, a codon-usage table is not a complete model of translation, and a sequence-distance matrix is not a complete evolutionary history. Quantitative molecular biology is strongest when it supports biological interpretation rather than replacing it.
In that sense, molecular biology is a model case of modern science: precise, data-rich, experimentally powerful, computationally analyzable, and yet always in need of interpretive connection across scales. The strongest molecular analysis connects sequence, expression, regulation, phenotype, environment, and history.
Why This Matters for Scientific Work
For working scientists, molecular biology matters because many biological problems are misread when the flow of genetic information is treated as a black box. A disease problem may depend on altered transcription rather than visible anatomy. A conservation problem may require understanding stress-response regulation rather than abundance alone. A microbial ecology problem may hinge on gene expression, horizontal transfer, or pathway activation. A plant or marine systems problem may require distinguishing inherited sequence variation from context-dependent expression response.
This means molecular biology should often be treated as explanatory infrastructure rather than as a narrow laboratory specialty. Ecologists need it because environmental response has molecular mechanisms. Evolutionary biologists need it because variation and adaptation have molecular substrates. Biomedical scientists need it because disease and therapy often depend on molecular pathways. Computational biologists need it because DNA, RNA, protein, expression matrices, and regulatory networks are core data structures of modern life science.
The scientific importance of molecular biology lies partly in this breadth. It is one of the principal ways biology explains how life persists, differentiates, responds, fails, adapts, and becomes knowable.
Molecular biology is also practically actionable. Sequences can be compared. Expression can be measured. Mutations can be detected. Transcripts can be quantified. Proteins can be inferred, modeled, and assayed. Molecular workflows can support diagnostics, ecological monitoring, biotechnology, conservation, pathogen tracking, plant breeding, and systems-level biological interpretation.
Conclusion
Molecular biology and the flow of genetic information show that living systems depend on the material organization, regulated transmission, and context-sensitive interpretation of hereditary information. DNA, RNA, and protein remain central not because biology can be reduced to them, but because they form one of the major axes through which continuity, function, adaptation, and change become scientifically intelligible.
To understand molecular biology is therefore to understand one of the deepest conditions of life: that information in living systems is stored materially, expressed selectively, altered historically, repaired imperfectly, and made functional through regulated molecular processes. That is why molecular biology remains central not only to genetics and cell biology, but also to ecology, conservation, microbiology, plant science, marine and freshwater biology, disease ecology, medicine, and biotechnology.
Molecular biology is thus more than a subfield. It is one of the principal ways modern biology understands how life persists, differentiates, adapts, and becomes knowable. Modern computational workflows deepen that understanding by making sequence comparison, transcript dynamics, expression change, codon usage, and molecular provenance more transparent, reproducible, and scientifically interpretable.
Related Articles
- Biology
- DNA, RNA, and the Molecular Logic of Life
- Genes, Inheritance, and the Principles of Heredity
- Cell Theory and the Basic Unit of Life
- Biomolecules and the Chemical Basis of Life
- Enzymes, Regulation, and Biochemical Pathways
- Metabolism, Energy, and Biological Function
- Cell Signaling, Communication, and Biological Coordination
- Mutation, Variation, and the Sources of Novelty
- Epigenetics, Regulation, and Gene Expression
- Genomics and the Expansion of Biological Knowledge
- Systems Biology and the Logic of Biological Integration
Further Reading
- Alberts, B. et al. (2002) Molecular Biology of the Cell. 4th edn. New York: Garland Science. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21054/
- Alberts, B. et al. (2002) ‘How cells read the genome: From DNA to protein’, in Molecular Biology of the Cell. 4th edn. New York: Garland Science. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21050/
- Alberts, B. et al. (2002) ‘From DNA to RNA’, in Molecular Biology of the Cell. 4th edn. New York: Garland Science. Available at: https://www.ncbi.nlm.nih.gov/books/NBK26887/
- Alberts, B. et al. (2002) ‘From RNA to protein’, in Molecular Biology of the Cell. 4th edn. New York: Garland Science. Available at: https://www.ncbi.nlm.nih.gov/books/NBK26829/
- Berg, J.M., Tymoczko, J.L. and Stryer, L. (2002) Biochemistry. 5th edn. New York: W.H. Freeman. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21154/
- Brown, T.A. (2002) ‘Genome replication’, in Genomes. 2nd edn. Oxford: Wiley-Liss. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21113/
- Brown, T.A. (2002) ‘Mutation, repair and recombination’, in Genomes. 2nd edn. Oxford: Wiley-Liss. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21114/
- Cooper, G.M. (2000) The Cell: A Molecular Approach. 2nd edn. Sunderland, MA: Sinauer Associates. Available at: https://www.ncbi.nlm.nih.gov/books/NBK9839/
- Koonin, E.V. and Galperin, M.Y. (2003) ‘Principles and methods of sequence analysis’, in Sequence – Evolution – Function: Computational Approaches in Comparative Genomics. Boston: Kluwer Academic. Available at: https://www.ncbi.nlm.nih.gov/books/NBK20261/
- Lodish, H. et al. (2000) Molecular Cell Biology. 4th edn. New York: W.H. Freeman. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21475/
- Mercadante, A.A. and Honda, B.M. (2023) ‘Biochemistry, replication and transcription’, in StatPearls. Treasure Island, FL: StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/NBK540152/
- Saeed, U. and Abbasi, B.A. (2019) ‘Biological sequence analysis’, in StatPearls. Treasure Island, FL: StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/NBK550342/
References
- Alberts, B. et al. (2002) Molecular Biology of the Cell. 4th edn. New York: Garland Science. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21054/
- Alberts, B. et al. (2002) ‘How cells read the genome: From DNA to protein’, in Molecular Biology of the Cell. 4th edn. New York: Garland Science. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21050/
- Alberts, B. et al. (2002) ‘From DNA to RNA’, in Molecular Biology of the Cell. 4th edn. New York: Garland Science. Available at: https://www.ncbi.nlm.nih.gov/books/NBK26887/
- Alberts, B. et al. (2002) ‘From RNA to protein’, in Molecular Biology of the Cell. 4th edn. New York: Garland Science. Available at: https://www.ncbi.nlm.nih.gov/books/NBK26829/
- Berg, J.M., Tymoczko, J.L. and Stryer, L. (2002) Biochemistry. 5th edn. New York: W.H. Freeman. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21154/
- Brown, T.A. (2002) ‘Genome replication’, in Genomes. 2nd edn. Oxford: Wiley-Liss. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21113/
- Brown, T.A. (2002) ‘Mutation, repair and recombination’, in Genomes. 2nd edn. Oxford: Wiley-Liss. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21114/
- Cooper, G.M. (2000) The Cell: A Molecular Approach. 2nd edn. Sunderland, MA: Sinauer Associates. Available at: https://www.ncbi.nlm.nih.gov/books/NBK9839/
- Koonin, E.V. and Galperin, M.Y. (2003) ‘Principles and methods of sequence analysis’, in Sequence – Evolution – Function: Computational Approaches in Comparative Genomics. Boston: Kluwer Academic. Available at: https://www.ncbi.nlm.nih.gov/books/NBK20261/
- Lodish, H. et al. (2000) Molecular Cell Biology. 4th edn. New York: W.H. Freeman. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21475/
- Mercadante, A.A. and Honda, B.M. (2023) ‘Biochemistry, replication and transcription’, in StatPearls. Treasure Island, FL: StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/NBK540152/
- Saeed, U. and Abbasi, B.A. (2019) ‘Biological sequence analysis’, in StatPearls. Treasure Island, FL: StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/NBK550342/