
Last Updated May 28, 2026
Mass spectrometry is one of chemistry’s most powerful methods for detecting molecules by turning them into ions and measuring their mass-to-charge behavior. It does not see neutral molecules directly. It transforms chemical substances into charged species, separates or analyzes those ions according to \(m/z\), records patterns of abundance, and allows chemists to infer molecular mass, formula, isotope composition, fragmentation behavior, compound identity, mixture composition, and trace-level presence. In modern chemistry, mass spectrometry sits at the intersection of ion chemistry, instrumentation, molecular detection, data science, and chemical inference.
The central thesis of this article is that a mass spectrum is not merely a list of peaks. It is a structured record of ion formation, mass analysis, detector response, calibration, fragmentation, isotope behavior, sample preparation, and computational interpretation. Molecular detection by mass spectrometry becomes scientifically meaningful only when ionization conditions, mass accuracy, resolution, abundance, adduct formation, fragmentation, reference data, uncertainty, and sample history are made visible.
Mass spectrometry is therefore both a measurement technology and a molecular evidence system. It can detect compounds at extremely low abundance, identify unknowns, quantify known analytes, characterize biomolecules, map metabolites, analyze polymers, support forensic and environmental investigations, and connect chemical structure to instrumental signal. But its strength depends on disciplined interpretation. A detected ion is not automatically a confirmed molecule, and an impressive peak is not automatically a reliable chemical conclusion.
Main Library
Publications
Article Map
Chemistry
Related Topic
Data Systems & Analytics
Related Topic
Biology
Related Topic
Environmental Monitoring Systems
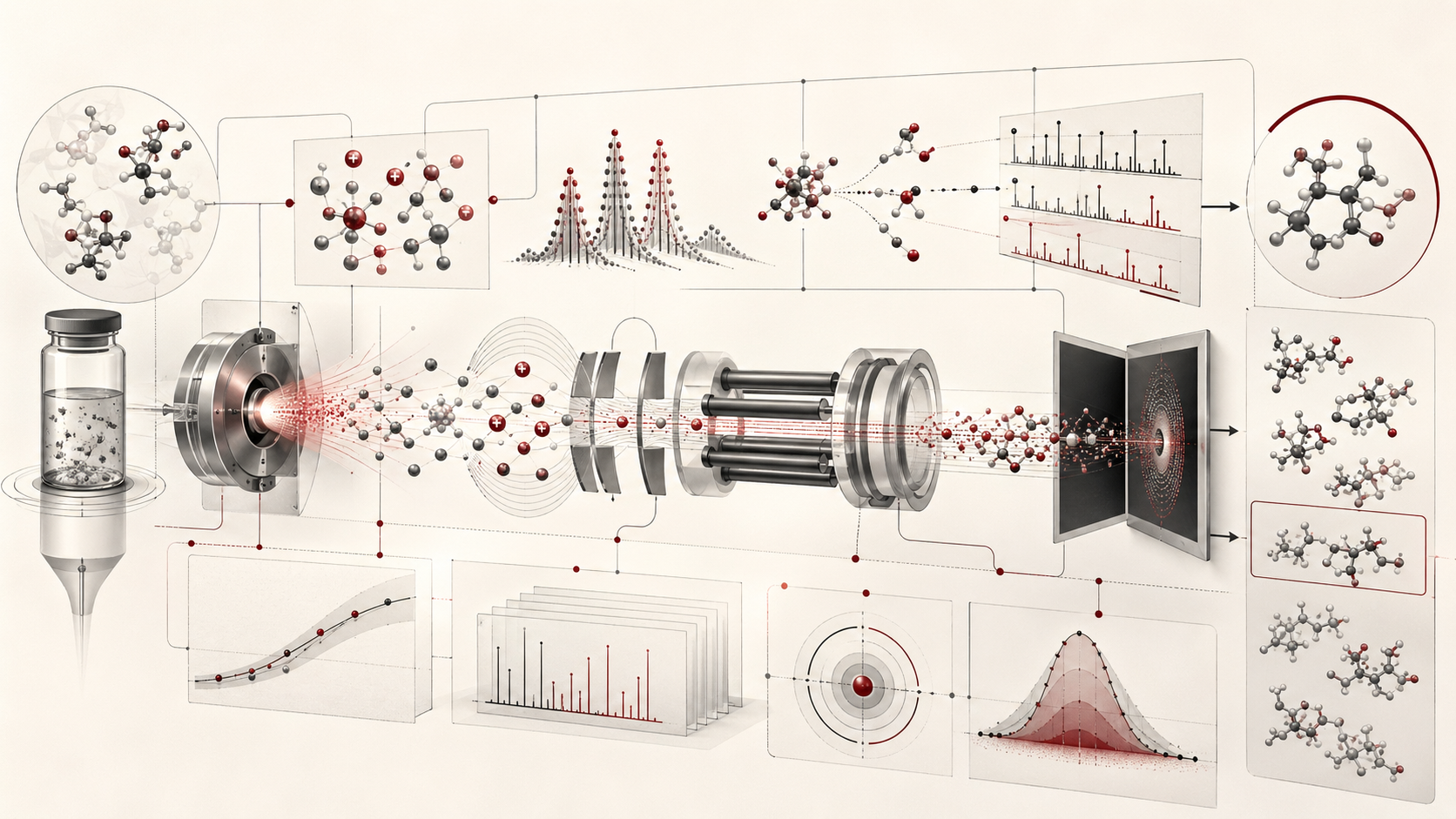
What Mass Spectrometry Measures
Mass spectrometry measures ions. A mass spectrum records signal intensity as a function of mass-to-charge behavior, conventionally written as \(m/z\). In chemistry, this matters because ions can carry information about the molecules from which they came. A protonated molecule, deprotonated molecule, radical cation, metal adduct, fragment ion, isotope peak, cluster ion, or multiply charged ion can provide evidence about molecular mass, elemental composition, structure, and chemical environment.
The phrase “molecular detection” must be used carefully. A mass spectrometer detects ions, not abstract molecular identities. A peak at a particular \(m/z\) may correspond to a molecular ion, an adduct, an isotope peak, a fragment, an in-source product, a contaminant, a background ion, a multiply charged species, a cluster, or a co-eluting compound. The act of identifying a molecule from mass spectrometric data is therefore inferential. It depends on mass accuracy, isotope pattern, retention time, fragmentation, reference spectra, chemical plausibility, sample history, and method validation.
This distinction is central to responsible interpretation. Mass spectrometry can be extraordinarily sensitive and specific, but a detected signal is not automatically a confirmed identity. Strong molecular detection requires a chain of evidence connecting sample preparation, ionization, mass analysis, signal processing, calibration, candidate generation, and confirmation.
Mass spectrometry also measures abundance imperfectly. Peak area or intensity can relate to amount, but response depends on ionization efficiency, matrix effects, detector behavior, chromatographic separation, adduct formation, source conditions, and instrument drift. Two compounds at the same concentration can produce very different signals. A compound can be abundant in a sample but ionize weakly, while another can be scarce but produce a strong response.
For researchers and scientists, mass spectrometry should be understood as a structured inference system. It measures ions, records signals, and supports molecular conclusions when chemical context, instrumental conditions, calibration, reference data, and uncertainty are handled explicitly.
Ions and Molecular Detection
Neutral molecules must be ionized before mass spectrometric analysis. Ionization can be gentle, producing intact molecular ions or adducts, or energetic, producing extensive fragmentation. These ion-formation conditions shape the spectrum. The same compound can produce different spectra under electron ionization, electrospray ionization, atmospheric pressure chemical ionization, matrix-assisted laser desorption/ionization, chemical ionization, photoionization, or other methods.
Common ion types include:
- Molecular ions: ions corresponding to the molecular mass of the analyte, often observed in electron ionization as radical cations.
- Protonated molecules: ions such as \([M+H]^+\), common in positive-mode electrospray and atmospheric-pressure ionization.
- Deprotonated molecules: ions such as \([M-H]^-\), common for acidic compounds in negative mode.
- Adduct ions: ions formed with sodium, potassium, ammonium, chloride, formate, acetate, lithium, solvent-related species, or buffer components.
- Fragment ions: ions formed by bond cleavage, rearrangement, neutral loss, or in-source fragmentation.
- Isotopic ions: peaks reflecting naturally occurring isotopes such as \(^{13}\mathrm{C}\), \(^{37}\mathrm{Cl}\), \(^{81}\mathrm{Br}\), \(^{34}\mathrm{S}\), and others.
- Multiply charged ions: ions carrying more than one charge, common in biomolecular mass spectrometry.
- Cluster ions: noncovalent or solvent-associated clusters that can appear under some source conditions.
Each ion type carries different evidence. A protonated molecule may support molecular-mass estimation. A fragment ion may support substructure inference. An isotope pattern may support elemental composition. A multiply charged envelope may support analysis of proteins, peptides, polymers, and large biomolecules. Molecular detection becomes stronger when these evidence types converge.
Ion chemistry also creates ambiguity. A sodium adduct may appear at a different \(m/z\) from a protonated molecule. In-source fragmentation may create a peak that looks like a separate compound. A background contaminant may appear in many samples. A doubly charged ion may appear at half the mass-to-charge value expected for a singly charged ion. A compound may produce different adducts in different solvents or instruments.
For researchers, the core interpretive discipline is to ask what ion was actually detected. A mass peak should be connected to ion type, charge state, adduct assumption, isotope pattern, fragmentation evidence, and sample context before it is treated as molecular identity.
Sample Preparation, Extraction, and Matrix Effects
Mass spectrometric evidence begins before the instrument. Sample collection, storage, extraction, cleanup, dilution, digestion, derivatization, desalting, filtration, centrifugation, solid-phase extraction, protein precipitation, solvent selection, and vial handling can all shape the final spectrum. A mass spectrometer may be sensitive, but it cannot correct for a poorly preserved, contaminated, degraded, or improperly extracted sample without appropriate controls.
Sample matrices matter because they influence ionization and detection. Blood, serum, urine, saliva, tissue, wastewater, soil extract, food, petroleum, polymer digest, atmospheric aerosol, and industrial process streams contain many species that can suppress or enhance ionization. Salts, detergents, phospholipids, proteins, humic substances, surfactants, polymers, buffers, acids, bases, and co-eluting compounds can all change response.
Matrix effects are especially important in electrospray and atmospheric-pressure ionization. A compound may ionize efficiently in pure solvent but poorly in a real sample. Another compound may be enhanced by matrix conditions. This is why matrix-matched calibration, standard addition, isotope-labeled internal standards, cleanup, chromatographic separation, and recovery studies are often essential for quantitative work.
Sample preparation also shapes identification. Derivatization can improve volatility, chromatographic behavior, ionization, or fragmentation, but it creates derivative-specific evidence. Digestion of proteins produces peptides rather than intact proteins. Hydrolysis, oxidation, photolysis, and microbial activity can change the analyte before measurement. Storage conditions can create degradation products or artifacts.
For researchers, sample preparation should be reported as part of the mass spectrometric method, not as a preliminary detail. A peak is evidence about the sample as prepared, extracted, introduced, ionized, and analyzed. Without preparation context, molecular interpretation weakens.
Ionization Methods and Chemical Context
Electron Ionization
Electron ionization is widely used in gas chromatography-mass spectrometry. It tends to produce reproducible fragmentation patterns for volatile and thermally stable compounds. Because many compounds fragment in characteristic ways under standardized conditions, electron ionization spectra can be compared with reference libraries. This makes GC-MS especially powerful for volatile organic compounds, solvents, fuels, environmental contaminants, forensic residues, flavor compounds, and many small molecules.
The strength of electron ionization is library comparability. Its limitation is that the molecular ion may be weak or absent for some compounds, and the method usually requires compounds that can enter the gas phase without decomposition. Thermally labile, highly polar, ionic, and large biomolecular compounds may require other approaches.
Chemical Ionization
Chemical ionization uses reagent ions to ionize analytes through ion-molecule reactions. It is often softer than electron ionization and can provide stronger molecular-ion-related signals. Positive and negative chemical ionization can be useful for confirming molecular mass, improving selectivity for certain compound classes, and reducing fragmentation.
Chemical ionization also introduces reagent-gas chemistry. The observed ions depend on reagent gas, pressure, ionization conditions, analyte proton affinity, electron affinity, and reaction pathways. Interpretation should state the reagent conditions and expected ion types.
Electrospray Ionization
Electrospray ionization is a soft ionization method widely used for polar, ionic, thermally labile, biological, and solution-phase molecules. It commonly produces protonated, deprotonated, adducted, and multiply charged ions. Because it can transfer large biomolecules from solution into the gas phase as multiply charged ions, electrospray is central to proteomics, metabolomics, lipidomics, pharmaceutical analysis, environmental analysis, and biomolecular chemistry.
Electrospray spectra are strongly shaped by solvent, salts, pH, mobile-phase additives, concentration, source conditions, and ion suppression. A compound’s presence in solution does not guarantee efficient ionization. Matrix effects can suppress or enhance signals, which is why internal standards, matrix-matched calibration, isotope dilution, and chromatographic separation are often needed.
Atmospheric Pressure Chemical Ionization and Related Methods
Atmospheric pressure chemical ionization and related methods are useful for less polar compounds that may not ionize efficiently by electrospray. These techniques are common in liquid chromatography-mass spectrometry workflows for pharmaceuticals, lipids, environmental compounds, and other small molecules. Ionization choice is therefore not a technical afterthought. It is a chemical decision about how a molecule is likely to become detectable.
MALDI and Desorption Ionization
Matrix-assisted laser desorption/ionization is often used for biomolecules, polymers, imaging, peptides, proteins, oligosaccharides, and materials. The analyte is embedded in a matrix that absorbs laser energy and assists ion formation. MALDI often produces singly charged ions even for large molecules, making spectra easier to interpret in some contexts. It can also support spatially resolved molecular imaging when applied to tissue or material surfaces.
MALDI results depend on matrix choice, crystallization, laser energy, sample preparation, salt content, surface uniformity, and local heterogeneity. In imaging work, spatial resolution and molecular assignment depend on tissue preparation, matrix deposition, calibration, and data-processing choices.
For researchers, ionization method should be chosen and reported as part of the chemical claim. Ionization determines what kinds of molecules are favored, what ions are likely, what evidence is produced, and what limitations remain.
Mass Analyzers and Instrument Architectures
After ions are formed, they must be separated or analyzed according to \(m/z\). Different mass analyzers use different physical principles and offer different tradeoffs in resolution, mass accuracy, speed, dynamic range, sensitivity, fragmentation capability, robustness, and cost. Instrument architecture shapes the evidence that can be produced.
Quadrupole Instruments
Quadrupole mass analyzers use oscillating electric fields to transmit ions within selected \(m/z\) windows. They are robust, widely used, and central to targeted quantification. Triple quadrupole instruments support selected reaction monitoring or multiple reaction monitoring, in which a precursor ion is selected, fragmented, and a characteristic product ion is monitored. This makes them powerful for quantitative assays in environmental chemistry, toxicology, pharmacokinetics, clinical research, food safety, and pharmaceutical analysis.
Quadrupole-based targeted methods are strong when analytes are known in advance and transitions are validated. They are weaker for unknown discovery because they usually monitor predefined ions. Their evidence strength depends on transition selection, retention time, ion ratios, calibration, matrix effects, and quality controls.
Time-of-Flight Instruments
Time-of-flight mass analyzers separate ions according to flight time after acceleration. Under appropriate conditions, ions with different \(m/z\) values reach the detector at different times. TOF instruments can offer high acquisition speed and high mass accuracy, making them useful for high-resolution mass spectrometry, GC-MS, LC-MS, MALDI, and non-targeted analysis.
TOF instruments are valuable when full-scan data, accurate mass, and rapid acquisition are important. Their interpretation still depends on calibration, resolving power, detector dynamic range, isotopic evidence, adduct logic, and fragmentation where needed.
Ion Traps
Ion traps confine ions using electric fields and can perform multiple stages of fragmentation. They are useful for structural analysis, tandem MS experiments, and workflows requiring sequential ion isolation and fragmentation. Their performance characteristics differ from quadrupole, TOF, Orbitrap, and Fourier-transform ion cyclotron resonance instruments.
Ion traps can provide rich fragmentation data, but ion storage, space-charge effects, low-mass cutoffs, isolation windows, and fragmentation conditions can influence spectra. Structural interpretation should preserve instrument settings and activation conditions.
Orbitrap and Fourier-Transform Instruments
Orbitrap and Fourier-transform ion cyclotron resonance instruments provide high resolving power and accurate mass measurement. They are central to high-resolution metabolomics, proteomics, petroleomics, environmental non-targeted analysis, isotope pattern interpretation, and molecular formula assignment. High resolving power can separate ions with very similar nominal masses, but high resolution alone does not guarantee correct identification.
Fourier-transform instruments are powerful for complex mixtures because they can distinguish closely spaced peaks. However, high-resolution data can create false confidence if mass accuracy is treated as identity proof. Formula assignment must still consider isotope pattern, adducts, retention behavior, fragmentation, chemical plausibility, and sample context.
Hybrid Instruments
Many modern systems combine analyzers. Quadrupole-time-of-flight instruments, quadrupole-Orbitrap instruments, ion trap-Orbitrap systems, and other hybrid designs allow precursor selection, fragmentation, accurate-mass detection, and high-resolution acquisition in one platform. These instruments support workflows that combine targeted quantification, suspect screening, and non-targeted discovery.
For researchers, instrument architecture should be matched to the analytical question. Targeted quantification, unknown identification, proteomics, imaging, isotope analysis, and polymer characterization may require different analyzer strengths. Instrument choice is part of the evidence design.
Fragmentation, Tandem MS, and Structural Evidence
Tandem mass spectrometry, often written MS/MS, adds structural information by isolating an ion, fragmenting it, and measuring the resulting product ions. In a typical workflow, a precursor ion is selected, activated by collision-induced dissociation, higher-energy collisional dissociation, electron-transfer dissociation, electron-capture dissociation, ultraviolet photodissociation, or another mechanism, and the fragments are analyzed.
Fragmentation is powerful because molecules do not break randomly. Fragment ions reflect bond strengths, functional groups, charge location, rearrangements, resonance stabilization, neutral losses, and ion chemistry. A loss of water may suggest hydroxyl or labile hydrogen environments. A loss of carbon dioxide may support carboxylate behavior. Peptide fragmentation can reveal sequence information. Lipid fragmentation can identify head groups, fatty acyl chains, or double-bond-related features under specialized methods.
However, fragmentation is not a simple structural code. The same nominal fragment can arise from different structures, and the same molecule can fragment differently under different collision energies, charge states, adducts, instruments, and acquisition conditions. Structural interpretation from MS/MS should therefore preserve the precursor ion, collision energy, ionization mode, product ions, intensities, reference spectra, and decision rules.
MS/MS evidence can support different levels of confidence. A transition in a targeted assay can support quantification when retention time, ion ratio, calibration, and quality controls are validated. A library match can support tentative identification when spectra are similar. A reference standard analyzed under the same conditions can provide stronger confirmation. A proposed structure based only on a few fragments may remain uncertain.
For researchers, tandem MS is strongest when used as part of convergent evidence: accurate precursor mass, isotope pattern, chromatographic retention, diagnostic fragments, spectral-library match, reference standard, blank control, and sample context. Fragmentation should strengthen inference, not replace careful interpretation.
Chromatography-Coupled Mass Spectrometry
Mass spectrometry is often coupled to chromatography because complex mixtures create overlapping ion signals, matrix effects, and ambiguous identifications. Gas chromatography-mass spectrometry separates volatile compounds before electron ionization and mass analysis. Liquid chromatography-mass spectrometry separates solution-phase compounds before atmospheric-pressure ionization and detection. Ion chromatography-MS, capillary electrophoresis-MS, supercritical-fluid chromatography-MS, and other coupled methods extend the same logic: separation reduces ambiguity before molecular detection.
Chromatography contributes retention evidence, reduces matrix effects, and helps distinguish isomers or coexisting compounds. Mass spectrometry contributes mass, isotope, and fragmentation evidence. Together, they create a stronger identification system than either technique alone. A compound detected by LC-MS/MS may be supported by retention time, accurate mass, isotope pattern, precursor ion, product ions, ion ratio, calibration behavior, and matrix-matched quality controls. In GC-MS, retention index and electron-ionization library matching are often used together.
Still, chromatography-coupled mass spectrometry requires caution. Co-elution can contaminate spectra. Matrix effects can distort abundance. In-source fragmentation can mimic product ions. Adduct formation can confuse molecular-mass interpretation. Library matches can be misleading if the library lacks relevant compounds or if spectra are low quality. Responsible identification requires structured evidence, not a single impressive peak.
Chromatographic conditions should therefore be reported clearly: column chemistry, dimensions, temperature, mobile phase or carrier gas, gradient, flow rate, injection volume, derivatization where relevant, retention-time windows, and quality-control behavior. Retention time is evidence only when the separation method is controlled.
For researchers, coupled methods should be interpreted as integrated systems. A retention time without mass evidence may be weak. A mass peak without separation may be ambiguous. A strong LC-MS or GC-MS claim explains how separation and mass evidence support each other.
Targeted, Suspect, and Non-Targeted Analysis
Mass spectrometry supports several modes of chemical investigation. Targeted analysis looks for predefined compounds using validated methods, standards, retention windows, transitions, calibration, and quality controls. Suspect screening searches for compounds from a candidate list without necessarily having standards for all of them. Non-targeted analysis attempts to detect and prioritize unknown features without prior knowledge of all possible compounds.
Targeted analysis is strongest for quantification and high-confidence reporting. It is used in pharmaceutical assays, environmental monitoring, toxicology, food safety, clinical research, and regulated testing. Its strength comes from defined analytes, known standards, calibration, method validation, and quality-control criteria.
Suspect screening is valuable when compounds of interest are plausible but not all standards are available. For example, environmental analysis may screen for transformation products, industrial chemicals, pharmaceuticals, or pesticide degradates. Suspect screening can use accurate mass, isotope pattern, retention-time prediction, MS/MS libraries, and chemical plausibility. However, identifications are often tentative unless confirmed with standards.
Non-targeted analysis is powerful for discovery but vulnerable to overinterpretation. It can reveal unexpected features, chemical fingerprints, exposure patterns, metabolomic shifts, or unknown contaminants. But feature detection, alignment, deconvolution, adduct grouping, blank subtraction, batch correction, and statistical filtering can all shape the result. Non-targeted workflows often generate many features that require prioritization and confidence grading.
For researchers, the reporting language should match the evidence mode. A targeted validated assay can report confirmed concentrations when criteria are satisfied. A suspect-screening result may report probable or tentative candidates. A non-targeted feature may remain an unresolved signal. Responsible mass spectrometry names uncertainty rather than hiding it.
Quantification, Internal Standards, and Calibration
Quantitative mass spectrometry connects signal to amount. The simplest approach uses external calibration standards, where peak area or height is modeled against known concentration. More robust methods often use internal standards, especially isotope-labeled analogs, to correct for extraction recovery, injection variability, ion suppression, and instrument drift.
Internal standards are powerful because they experience many of the same preparation and ionization conditions as the analyte. Isotope dilution mass spectrometry is especially strong when an isotopically labeled version of the analyte is added in known amount. The analyte and internal standard can be distinguished by mass while behaving similarly during extraction, chromatography, and ionization.
Quantitative evidence should include calibration range, weighting model, residuals, limits of detection and quantification, precision, bias, recovery, matrix effects, carryover, blank response, dilution integrity, stability, and quality-control acceptance criteria. A calibration curve with high \(R^2\) is not sufficient by itself. Calibration points may be unevenly weighted, low-concentration performance may be poor, and matrix effects may differ between standards and samples.
Quantification also depends on integration. Peak boundaries, baseline correction, smoothing, deconvolution, and co-elution can all change peak area. In targeted LC-MS/MS, ion ratios and retention times can detect some problems. In high-resolution workflows, extracted-ion chromatograms and isotope patterns can support quantitative interpretation. Still, automated integration should be reviewable.
For researchers, reported concentrations should be traceable to sample amount, extraction, dilution, calibration, internal standard response, matrix, QC status, and uncertainty. A concentration is the end of a measurement chain, not a raw instrument fact.
Isotopes, Adducts, Charge States, and Molecular Formula Inference
Isotopes provide powerful chemical evidence. Natural isotope patterns can reveal carbon count, chlorine or bromine presence, sulfur contribution, and charge state. A compound containing chlorine often shows a characteristic \(M+2\) pattern due to \(^{37}\mathrm{Cl}\). Bromine produces an especially distinctive pattern because \(^{79}\mathrm{Br}\) and \(^{81}\mathrm{Br}\) are both abundant. Carbon contributes to \(M+1\) through \(^{13}\mathrm{C}\). High-resolution isotope patterns can support molecular formula assignment.
Adducts can both help and complicate interpretation. A compound may appear as \([M+H]^+\), \([M+Na]^+\), \([M+K]^+\), \([M+NH_4]^+\), \([M-H]^-\), \([M+Cl]^-\), \([M+HCOO]^-\), or other adducts depending on ionization mode and solution composition. Adduct grouping can reveal that multiple peaks belong to the same neutral molecule. But incorrect adduct assumptions can lead to incorrect molecular masses and candidate formulas.
Charge-state determination is essential for multiply charged ions. Peptides, proteins, oligonucleotides, polymers, and large biomolecules often produce charge-state envelopes. Adjacent isotope peaks are separated approximately by \(1/z\), allowing charge estimation. Once charge is known, neutral mass can be inferred from \(m/z\) and adduct assumptions.
Molecular formula inference combines accurate mass, isotope pattern, charge state, adduct type, element constraints, and chemical plausibility. Many formulas can fit a mass within a narrow tolerance, especially as mass increases. Formula assignment should therefore be presented with confidence limits and supporting evidence rather than as automatic identity.
For researchers, isotope, adduct, and charge-state evidence should be integrated explicitly. A molecular formula candidate is stronger when exact mass, isotope pattern, adduct logic, charge state, fragmentation, retention behavior, and sample context are consistent.
Applications Across Chemistry, Biology, Environment, and Materials
Mass spectrometry is foundational across many fields because it can detect and characterize molecules in complex systems. In organic chemistry, it supports product confirmation, impurity detection, reaction monitoring, and structural analysis. In analytical chemistry, it supports trace detection, quantification, and method validation. In biochemistry, it supports proteomics, metabolomics, lipidomics, glycomics, and biomolecular characterization. In environmental chemistry, it supports detection of contaminants, transformation products, pollutants, and unknown features in water, air, soil, sediment, and biota.
In pharmaceutical and toxicological work, mass spectrometry supports drug metabolism, pharmacokinetics, impurity profiling, bioanalysis, forensic toxicology, anti-doping analysis, and quality control. In food chemistry, it supports contaminant detection, authenticity studies, flavor analysis, residue testing, and metabolite profiling. In materials chemistry, it can characterize polymers, additives, degradation products, surface species, volatile emissions, and complex organic mixtures.
Mass spectrometry also supports imaging and spatial chemistry. MALDI imaging and related methods can map molecular distributions across tissue, surfaces, materials, and biological structures. These workflows connect chemistry to spatial organization but require careful sample preparation, normalization, molecular assignment, and image interpretation.
For researchers, the application determines the evidence standard. A discovery metabolomics feature does not require the same confirmation as a forensic toxicology report. A preliminary polymer degradation screen does not require the same validation as a regulatory contaminant assay. A proteomics peptide-spectrum match does not prove the same thing as an isolated reference-standard match for a small molecule. The method should be judged by the decision it supports.
Data Standards, Spectral Libraries, and Reproducibility
Mass spectrometry produces complex data. Raw vendor files may contain profile spectra, centroided spectra, chromatograms, instrument methods, scan metadata, precursor isolation windows, collision energies, ion mobility values, calibration data, and processing histories. Reproducibility depends on preserving enough of this context to make interpretation auditable.
Open formats and community standards help. The mzML format, developed through the HUPO Proteomics Standards Initiative, provides a standard format for mass spectrometry output files. Such formats support data exchange, reanalysis, software interoperability, and long-term preservation. They do not solve every problem, but they reduce dependence on opaque vendor-specific data structures.
Spectral libraries are also central to molecular detection. Electron-ionization libraries support GC-MS identification. Tandem MS libraries support compound matching in LC-MS/MS, proteomics, metabolomics, lipidomics, and small-molecule analysis. NIST mass spectral resources are among the most important reference infrastructures for evaluated spectra and compound-identification workflows. However, library matching must be interpreted cautiously. A high match score can be misleading if the spectrum is contaminated, the compound is absent from the library, acquisition conditions differ, or the candidate is chemically implausible.
Good MS data practice includes:
- preserving raw data and converted open-format files where possible;
- recording ionization mode, source conditions, analyzer type, mass range, resolution, calibration, and acquisition method;
- recording chromatography conditions when MS is coupled to separation;
- tracking precursor ions, product ions, collision energies, isolation windows, and scan types in MS/MS workflows;
- documenting peak-picking, centroiding, smoothing, baseline correction, deconvolution, alignment, and normalization settings;
- stating whether identifications are tentative, probable, confirmed, unresolved, or reference-standard matched;
- linking reported candidates to reference data, fragment evidence, retention evidence, blanks, and quality-control results.
For researchers, reproducibility depends on preserving both data and interpretation. A raw file may be preserved, but if processing settings, library version, calibration record, and decision criteria are missing, the reported conclusion may still be difficult to reproduce.
Uncertainty, Quality Control, and False Identification
Mass spectrometry can produce false confidence because it offers precise-looking numbers. A peak at \(m/z = 301.1412\) may seem definitive, but many chemical interpretations may remain possible. Mass accuracy narrows the search space; it does not eliminate ambiguity. Fragmentation strengthens evidence; it does not always prove structure. Retention time supports identity; it can be shared by co-eluting compounds. Library matches are useful; they depend on data quality and library coverage.
Major sources of uncertainty include:
- mass calibration drift;
- ion suppression or enhancement;
- adduct formation;
- in-source fragmentation;
- co-elution;
- background contamination;
- carryover;
- poor peak integration;
- low signal-to-noise ratio;
- incorrect charge-state assignment;
- incorrect isotope or adduct grouping;
- insufficient library coverage;
- overinterpretation of exact mass alone.
Quality-control strategies include blanks, solvent blanks, matrix blanks, calibration standards, internal standards, isotope-labeled standards, quality-control pools, system suitability checks, replicate injections, retention-time windows, ion-ratio criteria, mass-error thresholds, signal-to-noise thresholds, pooled QC samples, and confirmatory MS/MS transitions. The exact controls depend on the application. Targeted pharmaceutical quantification, environmental non-target analysis, forensic toxicology, food-safety testing, proteomics, metabolomics, and materials analysis require different evidence standards.
False identification is especially important in non-targeted and suspect-screening workflows. Large feature tables can produce many plausible matches. Database search can return candidates that fit mass but are chemically irrelevant. Statistical significance can reflect batch effects rather than biology. A feature can be real without being identified, and an identified candidate can still be wrong.
For researchers, uncertainty should be reported rather than hidden. Identification confidence, mass error, retention evidence, MS/MS evidence, library score, blank presence, replicate reproducibility, and standard confirmation should be stated clearly enough for others to evaluate the claim.
Responsible Use of Mass Spectrometric Evidence
Mass spectrometry is used in high-consequence contexts: toxicology, environmental monitoring, forensic science, anti-doping, pharmaceutical quality, clinical research, food safety, homeland security, metabolomics, proteomics, and industrial process control. Responsible use requires clarity about the strength of identification and the limits of measurement.
Responsible MS practice includes:
- not treating exact mass alone as definitive molecular identity;
- distinguishing detected features from confirmed compounds;
- using reference standards when identity or quantification matters;
- documenting ionization mode, adduct assumptions, calibration, and mass tolerance;
- reporting whether identifications are tentative, probable, confirmed, unresolved, or reference-standard matched;
- using blanks and quality controls to detect contamination and carryover;
- preserving raw data, processing parameters, library versions, and audit trails;
- avoiding overstated claims in medical, forensic, environmental, and regulatory contexts.
Responsible use also includes communicating what mass spectrometry cannot prove alone. A compound detected in a sample does not automatically establish source, exposure route, toxicity, intent, causality, or biological effect. A metabolomic difference does not automatically reveal mechanism. A forensic detection does not automatically answer legal significance. A contaminant feature does not automatically establish regulatory exceedance without validated quantification and context.
The ethical strength of mass spectrometry lies not only in sensitivity, but in disciplined interpretation. A mass spectrum can detect astonishingly small chemical signals, but molecular detection becomes scientific evidence only when its ion chemistry, uncertainty, reference comparisons, and limitations are visible.
Mathematical Lens: \(m/z\), Mass Error, Resolution, Isotopes, and Calibration
The central measured quantity in mass spectrometry is conventionally written as \(m/z\). In simplified chemical reasoning, an ion with mass \(m\) and charge number \(z\) is observed according to:
m/z = \frac{m}{z}
\]
Interpretation: \(m/z\) describes mass-to-charge behavior. A peak’s observed value does not automatically reveal neutral molecular mass because the ion may be charged, adducted, fragmented, clustered, or isotopically substituted.
For a singly charged protonated molecule, neutral mass is often approximated as:
m_{\mathrm{neutral}} \approx (m/z)_{\mathrm{observed}} – m_{\mathrm{H}^{+}}
\]
Interpretation: This approximation assumes the observed ion is \([M+H]^+\). If the ion is a sodium adduct, potassium adduct, fragment, cluster, or multiply charged ion, a different calculation is required.
For a multiply charged adducted ion, a simplified neutral-mass estimate is:
m_{\mathrm{neutral}} \approx z(m/z)_{\mathrm{observed}} – z m_{\mathrm{adduct}}
\]
Interpretation: \(z\) is charge state and \(m_{\mathrm{adduct}}\) is the mass contribution per charge-carrying adduct. Correct charge-state and adduct assignment are essential.
Mass accuracy is often reported as mass error in parts per million:
\mathrm{ppm\ error} = \frac{m_{\mathrm{observed}} – m_{\mathrm{theoretical}}}{m_{\mathrm{theoretical}}} \times 10^{6}
\]
Interpretation: Small mass error can support candidate formulas, but it does not prove identity. Many formulas or isomers can share very similar masses, especially in complex chemical spaces.
Resolution is commonly expressed as:
R = \frac{m}{\Delta m}
\]
Interpretation: \(m\) is a selected mass and \(\Delta m\) is a measure of peak width, often at full width at half maximum depending on convention. Higher resolution can separate closer peaks and improve formula assignment, but it must be paired with calibration, signal quality, and chemical context.
Isotope spacing is especially useful for charge-state determination. For many isotope clusters, adjacent isotope peaks are separated approximately by:
\Delta(m/z) \approx \frac{1}{z}
\]
Interpretation: Isotope peaks spaced by about \(1.0\) suggest a singly charged ion, spacing near \(0.5\) suggests a doubly charged ion, and spacing near \(0.333\) suggests a triply charged ion.
For quantitative mass spectrometry, calibration models often connect peak area or peak height to concentration:
S_i = \beta_0 + \beta_1 c_i + e_i
\]
Interpretation: \(S_i\) is signal, \(c_i\) is concentration, \(\beta_0\) is intercept, \(\beta_1\) is response factor, and \(e_i\) is residual error. Internal standards and isotope dilution are often used to correct for extraction recovery, injection variability, ion suppression, and instrument drift.
A response ratio model with internal standard can be written as:
\frac{S_{\mathrm{analyte}}}{S_{\mathrm{IS}}} = \beta_0 + \beta_1 c_{\mathrm{analyte}} + e
\]
Interpretation: Internal-standard response can reduce variability from injection, extraction, and ionization, especially when the internal standard closely matches the analyte’s behavior.
These equations are useful because they make assumptions visible. Molecular mass depends on ion type. Mass error depends on calibration. Resolution depends on peak width. Charge state can be inferred from isotope spacing. Quantification depends on calibration, response, and matrix behavior. The equations structure interpretation; they do not replace chemical evidence.
Computational Workflows for Mass Spectrometry
Computational workflows can make mass spectrometric interpretation more transparent. A workflow can track sample identity, preparation method, instrument method, ionization mode, analyzer type, raw files, converted files, calibration status, feature detection settings, mass error, isotope pattern, adduct assignment, retention time, MS/MS evidence, library matches, blank flags, quality-control status, and identification confidence.
Useful workflows include exact-mass matching, isotope-spacing charge estimation, adduct grouping, blank subtraction, calibration-curve fitting, internal-standard correction, replicate abundance summaries, retention-time alignment, feature-table generation, suspect-list screening, spectral-library matching, false-discovery review, and molecular-evidence registers. More advanced workflows may integrate mzML files, vendor raw files, laboratory information systems, spectral libraries, molecular databases, cheminformatics, machine learning, and audit trails.
For researchers, computational workflows should preserve identification level. A feature should not automatically become a compound. A compound candidate should not automatically become a confirmed identification. A quantified concentration should not automatically become a regulatory conclusion. The workflow should carry confidence, evidence type, and limitations forward.
The examples below use synthetic data. They do not identify real compounds, validate analytical methods, support clinical or forensic reporting, or replace professional mass spectrometric interpretation. They demonstrate how MS reasoning can be structured, audited, and communicated responsibly.
Python Example: Mass Accuracy, Isotope Spacing, and Candidate Detection
The following Python example uses synthetic educational data to demonstrate mass-spectrometry reasoning. It calculates mass error in ppm, estimates charge state from isotope spacing, performs a simple candidate match, assigns tentative confidence, and writes outputs. It is intentionally simplified: real workflows require calibration records, adduct logic, retention time, isotope pattern scoring, MS/MS evidence, blank subtraction, quality controls, and expert review.
from pathlib import Path
from typing import Dict, List
import json
import pandas as pd
# Synthetic mass spectrometry workflow for molecular detection.
# Educational example only; not for clinical, forensic,
# environmental, pharmaceutical, or regulatory identification.
def calculate_ppm_error(observed_mz: float, theoretical_mz: float) -> float:
"""Return mass error in parts per million."""
return (observed_mz - theoretical_mz) / theoretical_mz * 1_000_000.0
def estimate_charge_from_isotope_spacing(spacing_mz: float) -> int:
"""Estimate charge state from isotope spacing."""
if spacing_mz <= 0:
raise ValueError("Isotope spacing must be positive.")
return int(round(1.0 / spacing_mz))
features = pd.DataFrame({
"feature_id": ["f1", "f2", "f3", "f4"],
"retention_time_min": [2.14, 3.88, 5.21, 7.45],
"observed_mz": [195.0878, 301.1412, 451.2129, 663.4521],
"charge": [1, 1, 2, 3],
"peak_area": [880000, 245000, 510000, 132000],
"blank_area": [1200, 800, 15000, 500],
})
candidate_library = pd.DataFrame({
"candidate": [
"caffeine_candidate",
"flavonoid_candidate",
"peptide_candidate",
"lipid_candidate",
],
"theoretical_mz": [195.08765, 301.14105, 451.21340, 663.45180],
"expected_charge": [1, 1, 2, 3],
"expected_rt_min": [2.10, 3.90, 5.25, 7.42],
"evidence_note": [
"synthetic exact-mass teaching candidate",
"synthetic exact-mass teaching candidate",
"synthetic multiply charged teaching candidate",
"synthetic multiply charged teaching candidate",
],
})
ppm_tolerance = 5.0
retention_tolerance_min = 0.20
blank_ratio_threshold = 0.05
matches: List[Dict[str, object]] = []
for _, feature in features.iterrows():
for _, candidate in candidate_library.iterrows():
if int(feature["charge"]) != int(candidate["expected_charge"]):
continue
ppm_error = calculate_ppm_error(
observed_mz=float(feature["observed_mz"]),
theoretical_mz=float(candidate["theoretical_mz"]),
)
rt_error = abs(
float(feature["retention_time_min"]) -
float(candidate["expected_rt_min"])
)
blank_ratio = float(feature["blank_area"]) / float(feature["peak_area"])
if abs(ppm_error) <= ppm_tolerance:
if rt_error <= retention_tolerance_min and blank_ratio < blank_ratio_threshold:
status = "tentative exact-mass and retention-time match"
else:
status = "tentative exact-mass match requiring review"
matches.append({
"feature_id": feature["feature_id"],
"candidate": candidate["candidate"],
"observed_mz": feature["observed_mz"],
"theoretical_mz": candidate["theoretical_mz"],
"ppm_error": ppm_error,
"retention_time_min": feature["retention_time_min"],
"retention_time_error_min": rt_error,
"peak_area": feature["peak_area"],
"blank_ratio": blank_ratio,
"identification_status": status,
})
match_table = pd.DataFrame(matches)
isotope_clusters = pd.DataFrame({
"cluster_id": ["c1", "c1", "c1", "c2", "c2", "c2"],
"isotope_peak": ["M", "M+1", "M+2", "M", "M+1", "M+2"],
"mz": [451.2130, 451.7132, 452.2131, 663.4520, 663.7855, 664.1186],
"relative_intensity": [1.00, 0.42, 0.12, 1.00, 0.61, 0.24],
})
charge_estimates: List[Dict[str, object]] = []
for cluster_id, group in isotope_clusters.groupby("cluster_id"):
group = group.sort_values("mz")
spacing = float(group["mz"].iloc[1] - group["mz"].iloc[0])
estimated_charge = estimate_charge_from_isotope_spacing(spacing)
charge_estimates.append({
"cluster_id": cluster_id,
"first_isotope_spacing_mz": spacing,
"estimated_charge": estimated_charge,
})
charge_table = pd.DataFrame(charge_estimates)
output_dir = Path("outputs")
output_dir.mkdir(exist_ok=True)
features.to_csv(output_dir / "synthetic_ms_features.csv", index=False)
match_table.to_csv(output_dir / "tentative_exact_mass_matches.csv", index=False)
charge_table.to_csv(output_dir / "isotope_charge_estimates.csv", index=False)
manifest: Dict[str, object] = {
"workflow": "synthetic_mass_spectrometry_detection",
"ppm_tolerance": ppm_tolerance,
"retention_tolerance_min": retention_tolerance_min,
"blank_ratio_threshold": blank_ratio_threshold,
"feature_count": int(len(features)),
"tentative_match_count": int(len(match_table)),
"responsible_use": [
"Exact mass alone is not definitive molecular identification.",
"Real workflows require calibration, isotope scoring, adduct logic, retention time, MS/MS, blanks, quality controls, reference standards, and expert review.",
],
}
with (output_dir / "mass_spectrometry_manifest.json").open(
"w",
encoding="utf-8"
) as file:
json.dump(manifest, file, indent=2)
print(match_table)
print(charge_table)The workflow demonstrates a fundamental principle: mass spectrometric detection becomes stronger when its assumptions are explicit. A candidate supported only by exact mass should be marked tentative. A candidate supported by exact mass, isotope pattern, retention time, MS/MS fragments, a reference standard, and quality controls can be reported with greater confidence.
R Example: Calibration, Replicate Abundance, and Detection Reporting
The following R example models a simple quantitative MS workflow using synthetic peak areas. It fits a calibration curve, estimates an unknown concentration, summarizes replicate measurements, and writes a report. In real analytical mass spectrometry, internal standards, matrix effects, recovery, instrument drift, uncertainty, and validated calibration models must be considered.
# Synthetic mass spectrometry calibration workflow.
# Educational example only; not for validated analytical reporting.
standards <- data.frame(
standard_id = c("blank", "std_01", "std_02", "std_03", "std_04", "std_05"),
concentration_ng_mL = c(0, 1, 5, 10, 25, 50),
analyte_peak_area = c(180, 18400, 90600, 181500, 452000, 905500),
internal_standard_area = c(250000, 248000, 252000, 251000, 249000, 250500)
)
unknowns <- data.frame(
sample_id = c("unknown_A", "unknown_A", "unknown_A"),
injection_id = c("inj_01", "inj_02", "inj_03"),
analyte_peak_area = c(327500, 331200, 325900),
internal_standard_area = c(249500, 251000, 248800)
)
standards$response_ratio <-
standards$analyte_peak_area / standards$internal_standard_area
unknowns$response_ratio <-
unknowns$analyte_peak_area / unknowns$internal_standard_area
calibration_model <- lm(
response_ratio ~ concentration_ng_mL,
data = standards
)
intercept <- coef(calibration_model)[1]
slope <- coef(calibration_model)[2]
unknowns$estimated_concentration_ng_mL <-
(unknowns$response_ratio - intercept) / slope
summary_table <- data.frame(
sample_id = "unknown_A",
mean_analyte_peak_area = mean(unknowns$analyte_peak_area),
sd_analyte_peak_area = sd(unknowns$analyte_peak_area),
mean_response_ratio = mean(unknowns$response_ratio),
sd_response_ratio = sd(unknowns$response_ratio),
mean_concentration_ng_mL =
mean(unknowns$estimated_concentration_ng_mL),
sd_concentration_ng_mL =
sd(unknowns$estimated_concentration_ng_mL),
replicate_count = nrow(unknowns)
)
summary_table$precision_review_required <-
summary_table$sd_concentration_ng_mL /
summary_table$mean_concentration_ng_mL > 0.15
dir.create("outputs", showWarnings = FALSE)
write.csv(
standards,
file = "outputs/ms_calibration_standards.csv",
row.names = FALSE
)
write.csv(
unknowns,
file = "outputs/ms_unknown_estimates.csv",
row.names = FALSE
)
write.csv(
summary_table,
file = "outputs/ms_quant_summary.csv",
row.names = FALSE
)
sink("outputs/ms_calibration_report.txt")
cat("Synthetic Mass Spectrometry Calibration Report\n")
cat("=============================================\n\n")
cat("Calibration model using response ratio:\n")
print(summary(calibration_model))
cat("\nUnknown sample summary:\n")
print(summary_table)
cat("\nResponsible-use note:\n")
cat("Synthetic educational data only. Real quantitative MS requires validated calibration, internal standards, matrix-effect assessment, blanks, quality controls, uncertainty analysis, and documented sample preparation.\n")
sink()
print(summary_table)This workflow illustrates why quantitative mass spectrometry is not merely peak measurement. It is a system of calibration, sample preparation, ionization control, detector response, replicate measurement, internal-standard correction, and uncertainty reporting. The reported concentration is the end of a measurement chain, not an isolated number.
SQL Example: Mass Spectrometry Evidence Register
Mass spectrometric interpretation becomes more reliable when sample preparation, instrument method, raw files, features, candidates, fragments, calibration, QC checks, and identification confidence are traceable. A simple evidence register can preserve the context needed to audit molecular detection claims.
CREATE TABLE ms_sample (
sample_id TEXT PRIMARY KEY,
sample_name TEXT NOT NULL,
sample_matrix TEXT,
collection_datetime TEXT,
storage_condition TEXT,
preparation_method TEXT,
preparation_notes TEXT
);
CREATE TABLE ms_instrument_method (
method_id TEXT PRIMARY KEY,
method_name TEXT NOT NULL,
method_version TEXT,
ionization_mode TEXT,
ion_source TEXT,
mass_analyzer TEXT,
chromatography_method TEXT,
acquisition_mode TEXT,
mass_range TEXT,
calibration_status TEXT,
method_notes TEXT
);
CREATE TABLE ms_run (
run_id TEXT PRIMARY KEY,
sample_id TEXT NOT NULL,
method_id TEXT NOT NULL,
injection_datetime TEXT,
raw_file_uri TEXT,
converted_file_uri TEXT,
file_checksum TEXT,
operator_or_scheduler TEXT,
run_status TEXT,
FOREIGN KEY (sample_id) REFERENCES ms_sample(sample_id),
FOREIGN KEY (method_id) REFERENCES ms_instrument_method(method_id)
);
CREATE TABLE ms_feature (
feature_id TEXT PRIMARY KEY,
run_id TEXT NOT NULL,
retention_time_min REAL CHECK (retention_time_min >= 0),
observed_mz REAL CHECK (observed_mz >= 0),
charge INTEGER CHECK (charge >= 1),
peak_area REAL CHECK (peak_area >= 0),
signal_to_noise REAL CHECK (signal_to_noise >= 0),
blank_flag INTEGER CHECK (blank_flag IN (0, 1)),
feature_quality_flag TEXT,
FOREIGN KEY (run_id) REFERENCES ms_run(run_id)
);
CREATE TABLE ms_candidate_identification (
candidate_id INTEGER PRIMARY KEY,
feature_id TEXT NOT NULL,
candidate_name TEXT,
candidate_formula TEXT,
theoretical_mz REAL CHECK (theoretical_mz >= 0),
adduct_assignment TEXT,
ppm_error REAL,
isotope_match_score REAL CHECK (isotope_match_score BETWEEN 0 AND 1),
retention_evidence TEXT,
library_match_score REAL CHECK (library_match_score BETWEEN 0 AND 1),
identification_level TEXT,
identification_notes TEXT,
FOREIGN KEY (feature_id) REFERENCES ms_feature(feature_id)
);
CREATE TABLE ms_fragment_evidence (
fragment_id INTEGER PRIMARY KEY,
feature_id TEXT NOT NULL,
precursor_mz REAL CHECK (precursor_mz >= 0),
product_mz REAL CHECK (product_mz >= 0),
product_intensity REAL CHECK (product_intensity >= 0),
collision_energy TEXT,
fragment_assignment TEXT,
FOREIGN KEY (feature_id) REFERENCES ms_feature(feature_id)
);
CREATE TABLE ms_quant_calibration (
calibration_id INTEGER PRIMARY KEY,
method_id TEXT NOT NULL,
analyte_name TEXT,
calibration_model TEXT,
concentration_range TEXT,
response_factor REAL,
intercept REAL,
r_squared REAL CHECK (r_squared BETWEEN 0 AND 1),
internal_standard TEXT,
limit_of_detection REAL CHECK (limit_of_detection >= 0),
limit_of_quantification REAL CHECK (limit_of_quantification >= 0),
calibration_quality_flag TEXT,
FOREIGN KEY (method_id) REFERENCES ms_instrument_method(method_id)
);
CREATE TABLE ms_quality_control (
qc_id INTEGER PRIMARY KEY,
run_id TEXT NOT NULL,
qc_type TEXT,
qc_status TEXT,
expected_value REAL,
measured_value REAL,
acceptance_min REAL,
acceptance_max REAL,
qc_notes TEXT,
FOREIGN KEY (run_id) REFERENCES ms_run(run_id)
);
SELECT
f.feature_id,
r.sample_id,
m.ionization_mode,
m.mass_analyzer,
f.retention_time_min,
f.observed_mz,
f.charge,
f.peak_area,
f.signal_to_noise,
c.candidate_name,
c.adduct_assignment,
c.ppm_error,
c.isotope_match_score,
c.library_match_score,
c.identification_level,
q.qc_status,
CASE
WHEN q.qc_status IS NOT NULL AND q.qc_status != 'pass'
THEN 'quality control review required'
WHEN f.blank_flag = 1
THEN 'blank contamination review required'
WHEN f.signal_to_noise < 10
THEN 'low signal review required'
WHEN ABS(c.ppm_error) > 5
THEN 'mass accuracy review required'
WHEN c.identification_level IN ('exact mass only', 'tentative')
THEN 'identification confidence review required'
ELSE 'standard review'
END AS ms_review_status
FROM ms_feature f
JOIN ms_run r
ON f.run_id = r.run_id
JOIN ms_instrument_method m
ON r.method_id = m.method_id
LEFT JOIN ms_candidate_identification c
ON f.feature_id = c.feature_id
LEFT JOIN ms_quality_control q
ON r.run_id = q.run_id
ORDER BY ms_review_status, f.retention_time_min;The purpose of this register is to keep mass spectrometric interpretation attached to evidence. A molecular identification should preserve sample identity, method version, raw data, ionization mode, analyzer type, feature information, candidate match, mass error, isotope evidence, fragment evidence, retention evidence, QC status, and confidence level. Mass spectrometry data become stronger when provenance is part of the record.
GitHub Repository
The companion repository for this article can support reproducible workflows for exact-mass matching, isotope-spacing charge estimation, calibration modeling, internal-standard correction, replicate abundance summaries, candidate evidence tracking, SQL provenance, and responsible MS interpretation.
Complete Code Repository
The full code distribution for this article, including selected mass spectrometry examples, expanded computational workflows, reproducible data structures, provenance documentation, candidate-detection scripts, calibration summaries, isotope-charge estimates, SQL evidence registers, and scientific-computing scaffolding, is available on GitHub.
Limits, Uncertainty, and Responsible Interpretation
Mass spectrometry is powerful, but it is not self-interpreting. A mass spectrum can suggest molecular identity, but the strength of that suggestion depends on evidence quality. Exact mass, isotope pattern, retention time, fragmentation, library match, reference standard, and quality controls all contribute different kinds of support. None should be confused with certainty when used alone.
Ionization introduces selectivity. Some compounds ionize efficiently; others do not. Some form multiple adducts. Some fragment in-source. Some are suppressed by matrix components. A compound absent from the spectrum is not necessarily absent from the sample. A compound with a large peak is not necessarily the most abundant neutral compound in the original material.
Data processing introduces additional uncertainty. Peak picking, centroiding, smoothing, baseline subtraction, deconvolution, feature alignment, blank subtraction, library scoring, molecular formula generation, and statistical filtering can all change results. Processing parameters should therefore be preserved and reviewed, especially in non-targeted and high-consequence workflows.
Identification confidence should match the evidence. A feature with accurate mass only should remain tentative. A feature with accurate mass and MS/MS library match may be more probable. A compound confirmed with a reference standard under the same method can support stronger identification. A quantitative report requires validated calibration and quality controls. A forensic, clinical, environmental, or regulatory conclusion requires method standards appropriate to that domain.
The computational examples associated with this article are synthetic and educational. They do not identify real compounds, validate analytical methods, certify clinical or forensic findings, establish environmental compliance, determine regulatory suitability, or replace professional mass spectrometry review. They are designed to show how mass-spectrometric reasoning can be structured and audited.
Responsible interpretation should avoid both instrument worship and instrument skepticism. Mass spectrometry can detect molecular evidence with extraordinary sensitivity, but its conclusions remain scientific claims that require context, controls, uncertainty, and disciplined reporting.
Conclusion
Mass spectrometry shows how chemistry can infer molecular identity from charged particles. Neutral molecules are transformed into ions, ions are separated by mass-to-charge behavior, signals are recorded, and chemists interpret patterns of mass, abundance, isotope structure, fragmentation, retention behavior, and calibration response. This makes mass spectrometry one of the most powerful forms of molecular evidence in modern chemistry.
The field’s central lesson is that a mass spectrum is not merely a peak list. It is the outcome of sample preparation, ionization, mass analysis, detector response, data processing, reference comparison, and chemical reasoning. A peak becomes evidence only when the path from sample to ion to spectrum to interpretation is visible.
For chemistry as a discipline, mass spectrometry is essential because it supports molecular detection across organic chemistry, analytical chemistry, biochemistry, environmental science, pharmacology, toxicology, food chemistry, forensic science, materials chemistry, and systems biology. It can reveal what is present, what is changing, what is contaminating, what is fragmenting, what is metabolizing, and what remains unknown.
A mature mass spectrometric practice does not ask only, “Is there a peak?” It asks: What ion produced the peak? What evidence supports the candidate? What else could explain the signal? What controls were used? What uncertainty remains? What decision will this interpretation support? The reliability of molecular detection depends on answering those questions with scientific discipline.
Related articles
- What Is Chemistry?
- Measurement, Quantification, and the Experimental Basis of Chemistry
- Chemical Metrology, Standards, and Reference Materials
- Mathematics for Chemistry and Molecular Systems
- Computational Notebooks and Reproducible Chemical Research
- Spectroscopy and the Measurement of Molecular Structure
- Chromatography, Separation Science, and Chemical Identification
- Electroanalytical Chemistry and Chemical Sensors
- Laboratory Automation, Chemical Data, and Instrument Workflows
- Analytical Chemistry and the Identification of Matter
- Environmental Chemistry and the Chemical Conditions of Habitability
- Biochemistry and the Molecular Basis of Life
Further reading
- de Hoffmann, E. and Stroobant, V. (2007) Mass Spectrometry: Principles and Applications. 3rd edn. Chichester: Wiley.
- Gross, J.H. (2017) Mass Spectrometry: A Textbook. 3rd edn. Cham: Springer.
- Harris, D.C. (2020) Quantitative Chemical Analysis. 10th edn. New York: W.H. Freeman.
- International Union of Pure and Applied Chemistry (n.d.) Mass-to-Charge Ratio in Mass Spectrometry. Available at: https://goldbook.iupac.org/terms/view/M03752
- HUPO Proteomics Standards Initiative (n.d.) mzML. Available at: https://www.psidev.info/mzml
- Martens, L. et al. (2010) ‘mzML—a Community Standard for Mass Spectrometry Data’, Molecular & Cellular Proteomics. Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3013463/
- National Institute of Standards and Technology (n.d.) Mass Spectrometry Data Center. Available at: https://chemdata.nist.gov/
- National Institute of Standards and Technology (n.d.) NIST Chemistry WebBook. Available at: https://webbook.nist.gov/chemistry/
- National Institute of Standards and Technology (n.d.) Tandem Mass Spectral Library. Available at: https://www.nist.gov/programs-projects/tandem-mass-spectral-library
- Watson, J.T. and Sparkman, O.D. (2007) Introduction to Mass Spectrometry: Instrumentation, Applications, and Strategies for Data Interpretation. 4th edn. Chichester: Wiley.
References
- de Hoffmann, E. and Stroobant, V. (2007) Mass Spectrometry: Principles and Applications. 3rd edn. Chichester: Wiley.
- Gross, J.H. (2017) Mass Spectrometry: A Textbook. 3rd edn. Cham: Springer.
- Harris, D.C. (2020) Quantitative Chemical Analysis. 10th edn. New York: W.H. Freeman.
- HUPO Proteomics Standards Initiative (n.d.) mzML. Available at: https://www.psidev.info/mzml
- International Union of Pure and Applied Chemistry (n.d.) Mass-to-Charge Ratio in Mass Spectrometry. Available at: https://goldbook.iupac.org/terms/view/M03752
- Martens, L. et al. (2010) ‘mzML—a Community Standard for Mass Spectrometry Data’, Molecular & Cellular Proteomics, 10(1). Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3013463/
- Murray, K.K. et al. (2013) ‘Definitions of terms relating to mass spectrometry’, Pure and Applied Chemistry, 85(7), pp. 1515–1609. Available at: https://publications.iupac.org/pac/pdf/2013/pdf/8507×1515.pdf
- National Institute of Standards and Technology (n.d.) Mass Spectrometry Data Center. Available at: https://chemdata.nist.gov/
- National Institute of Standards and Technology (n.d.) NIST Chemistry WebBook. Available at: https://webbook.nist.gov/chemistry/
- National Institute of Standards and Technology (n.d.) Tandem Mass Spectral Library. Available at: https://www.nist.gov/programs-projects/tandem-mass-spectral-library
- Skoog, D.A., Holler, F.J. and Crouch, S.R. (2017) Principles of Instrumental Analysis. 7th edn. Boston: Cengage Learning.
- Wallace, W.E. (2023) ‘NIST Mass Spectrometry Data Center, Standard Reference Data, and Software’, Journal of the American Society for Mass Spectrometry. Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC10517720/
- Watson, J.T. and Sparkman, O.D. (2007) Introduction to Mass Spectrometry: Instrumentation, Applications, and Strategies for Data Interpretation. 4th edn. Chichester: Wiley.