
Last Updated May 28, 2026
Epigenetics, regulation, and gene expression examine how living systems control which genes are active, when they are active, where they are active, and to what extent they are expressed across cells, tissues, organisms, populations, and environments. Epigenetics is central to modern biology because DNA sequence alone does not explain how genetically similar cells become different cell types, how organisms respond to environmental conditions, how developmental states persist, or how biological systems stabilize and modify patterns of activity across time. A genome is not a self-reading script. It is a structured biochemical substrate interpreted through chromatin organization, transcription-factor binding, RNA-mediated control, nuclear architecture, signaling context, metabolic state, developmental history, and environmental condition.
This article develops Epigenetics, Regulation, and Gene Expression as a foundational article within the Biology knowledge series. It treats gene regulation not as a decorative layer added after genetics, but as one of the core systems through which biological information becomes function, identity, plasticity, disease, and environmental response. Genes matter because they provide inherited sequence and functional possibility. Regulation matters because it determines which possibilities become active in particular cells, tissues, life stages, and environmental contexts.
Main Library
Publications
Article Map
Biology
Related Topic
Chemistry
Related Topic
Earth Science
Related Topic
Environmental Science
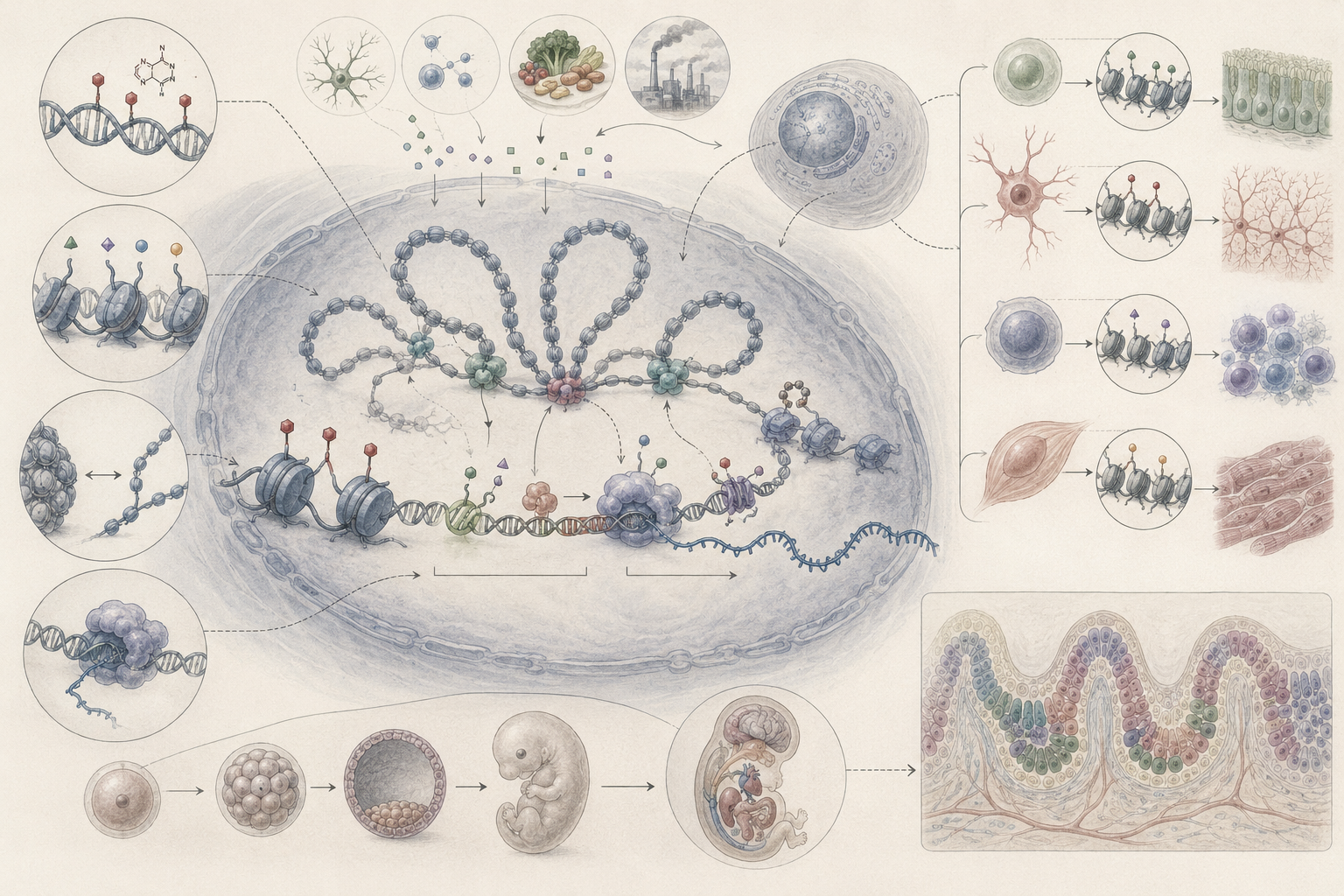
The article develops epigenetics, regulation, and gene expression across chromatin organization, DNA methylation, histone modification, regulatory RNA, transcriptional control, RNA turnover, translation control, cell-state memory, differentiation, development, physiology, ecology, medicine, biotechnology, computational epigenomics, single-cell biology, plant and microbial stress response, disease dysregulation, and systems-level biological organization.
The article also extends epigenetic biology into quantitative and computational analysis through transcript-decay modeling, production-decay dynamics, two-state regulatory switching, methylation fractions, accessibility scoring, differential expression, regulatory concordance screening, Markov-style cell-state transitions, R workflows, Python workflows, SQL provenance structures, and a linked full-stack GitHub repository containing Python, R, Julia, Fortran, Rust, Go, C, C++, SQL, notebooks, data files, validation notes, and reproducibility documentation.
What epigenetics and gene expression are
Gene expression is the process by which genetic information becomes biologically active through transcription, RNA processing, translation, and related regulatory steps. Epigenetics refers to systems of biological regulation that influence gene activity without requiring a change in the underlying DNA sequence itself. In broad terms, epigenetics helps explain how the same genome can support many different cellular, developmental, physiological, and environmental states.
This distinction matters because biology cannot be understood through sequence alone. Genes may be present in a genome, yet not all are active at the same time, in the same tissue, or under the same conditions. Some genes are strongly expressed in one cell type and silenced in another. Some become active only in response to environmental stress, developmental signaling, nutrient state, immune challenge, toxic exposure, pathogen pressure, or disease process. Epigenetics belongs to the study of how these differences are organized, stabilized, modulated, and sometimes remembered.
Epigenetics and gene expression therefore sit at the center of modern biology. They link DNA to phenotype, heredity to development, regulation to function, and environment to biological response. Without them, the genome would remain an insufficient explanation for how living systems actually work. A serious understanding of biology requires not only knowing what genes exist, but how cells decide which programs to enact, suppress, delay, amplify, localize, degrade, or remember.
Epigenetics is also important because it helps biology avoid two opposite errors. The first error is genetic determinism: the idea that sequence alone directly and completely determines biological outcome. The second error is treating regulation as vague or mystical, detached from molecular mechanism. Modern epigenetics does neither. It studies concrete biological systems—chromatin, methylation, histone marks, regulatory proteins, RNA, nuclear organization, and cell-state memory—while preserving the broader insight that biological meaning depends on context.
Why DNA sequence alone is not enough
DNA sequence is foundational, but it is not self-interpreting. A genome does not automatically explain which genes are active in a neuron versus a muscle cell, why a seed remains dormant before germinating, how a plant responds to drought, how immune cells rapidly change state under infection, or why one cellular lineage preserves identity while another differentiates. These outcomes depend not only on what information is present, but on how it is accessed, regulated, transcribed, processed, translated, stabilized, and coordinated across time.
This is one reason epigenetics became so important. It helped biology move beyond the mistaken idea that the genome is a fixed script passively read in the same way everywhere. Instead, it revealed that genomes are interpreted through structured states of accessibility, modification, repression, activation, and memory. Cells are not simply different because they contain different genes. They are often different because they regulate shared genes differently.
That insight has major consequences. It means biological identity depends not only on sequence inheritance, but also on regulatory context. It also means environmental influence can become biologically consequential through changes in gene activity, chromatin state, regulatory architecture, and RNA dynamics without necessarily requiring a new mutation. Sequence supplies the informational substrate, but regulation determines which fractions of that substrate become functionally operative in real cells and real environments.
This is especially important in multicellular organisms, where many cell types share nearly identical DNA but behave in radically different ways. A neuron, hepatocyte, macrophage, root hair cell, guard cell, pollen grain, fungal spore, or intestinal epithelial cell may differ less because of sequence content than because of regulatory state. Gene expression explains how shared inherited information becomes specialized biological identity.
Chromatin and the accessibility of the genome
One of the most important principles in epigenetics is that DNA is packaged into chromatin, and that chromatin state influences whether genomic regions are accessible or relatively silenced. DNA is not left exposed as a naked informational string inside the nucleus. It is wrapped around histone proteins and organized into higher-order structures that affect transcriptional access, replication behavior, repair, nuclear positioning, and regulatory control.
This matters because accessibility is one of the first conditions of expression. A gene region that is tightly compacted is less likely to be actively transcribed than one in a more open and accessible chromatin state. Epigenetics is therefore deeply connected to the physical organization of the genome. Regulation is not only a matter of transcription factors floating through cells. It is also a matter of whether DNA is structurally available to be read.
Chromatin biology thus changed the logic of heredity and expression. It showed that the genome is both a sequence system and an architectural system, and that gene activity depends partly on how those two levels are coordinated. Euchromatin, heterochromatin, enhancer accessibility, nucleosome positioning, local compaction, chromatin remodeling, and three-dimensional folding all influence the regulatory landscape. In modern biology, gene control is inseparable from genome topology and structural context.
Chromatin is also dynamic. It changes across development, cell differentiation, disease states, stress response, immune activation, and environmental exposure. A regulatory region may become accessible during a developmental window and then close later. A stress-response locus may open transiently under environmental pressure. A cancer cell may alter chromatin architecture in ways that destabilize identity and activate abnormal programs. Chromatin is therefore not merely packaging. It is one of the principal media through which cells interpret and reorganize genomic possibility.
DNA methylation, histone modification, and epigenetic marking
DNA methylation and histone modification are among the best-known epigenetic mechanisms because they influence the activity of genomic regions without changing nucleotide sequence. DNA methylation is often associated with altered transcriptional potential, frequently repression in many contexts, while histone modifications can influence whether local chromatin becomes more open, more compact, or differentially interpreted by regulatory machinery.
These mechanisms matter because they provide cells with ways to stabilize patterns of activity. A cell type does not need to rediscover its identity at every moment from scratch. It can maintain differentiated states, silence certain programs, preserve functional specialization, and respond selectively to signals through epigenetic regulation. This is especially important in development, where long-lived cellular states emerge from initially more plastic conditions.
At the same time, epigenetic marks are not merely static labels pasted onto the genome. Many are dynamic, context-sensitive, reversible, and biologically responsive. Their significance lies in the regulated control of access, timing, and transcriptional possibility rather than in any one mark considered in isolation. Modern chromatin biology emphasizes combinatorial logic: multiple marks, chromatin remodelers, DNA-binding proteins, RNA molecules, nuclear context, and local sequence features interact to produce regulatory states that are probabilistic, layered, and historically conditioned.
This is why epigenetic interpretation requires caution. A methylation mark, histone modification, or accessibility signal may correlate with expression, but correlation is not always causation. Some marks help establish regulatory state, some help maintain it, some reflect it, and some participate differently depending on cell type or locus. The strongest epigenetic claims connect molecular marks to mechanism, expression, perturbation, phenotype, and biological context.
Regulatory RNA and post-transcriptional control
Epigenetics and gene regulation also extend beyond chromatin. Regulatory RNA molecules play major roles in silencing, fine-tuning, stabilizing, degrading, localizing, or otherwise shaping gene expression. Small RNAs, microRNAs, siRNAs, piRNAs, long noncoding RNAs, enhancer RNAs, antisense transcripts, and other RNA-based mechanisms contribute to the control of transcriptional state, RNA turnover, translation, and genomic regulation. This expands the picture considerably because it means that gene control is not confined to DNA-protein interactions alone.
Post-transcriptional regulation is equally important because expression is not determined solely at the moment of transcription. Messenger RNAs may be spliced differently, degraded at different rates, translated with different efficiencies, edited, sequestered, localized, or released according to cellular state. Gene expression therefore unfolds through multiple checkpoints rather than through one single on/off event.
This broader framework is one reason modern biology treats gene regulation as layered. Sequence, chromatin, RNA, protein factors, cellular state, metabolism, signaling, and environmental context all interact to determine what genes actually do in living systems. Regulatory RNA is particularly important because it reveals how expression can be tuned continuously rather than merely permitted or denied. Fine control of dosage, timing, localization, and turnover is often as biologically important as the original act of transcription.
RNA regulation is also central to stress response and disease. A cell may respond rapidly to heat, hypoxia, infection, nutrient limitation, toxin exposure, or inflammation by changing RNA stability and translation before longer-term transcriptional changes fully unfold. In this sense, post-transcriptional control helps explain how living systems achieve both speed and specificity.
Gene expression as a dynamic biological process
Gene expression is best understood dynamically rather than statically. Genes may pulse, oscillate, switch, rise gradually, respond transiently, or remain stably active over long periods. Some expression changes are rapid and reversible, while others are developmentally durable or physiologically persistent. Expression therefore belongs not only to molecular biology, but also to systems biology and time-dependent biological analysis.
This dynamic view matters because many biological functions depend on timing as much as on presence. A transcription factor expressed too early, too late, too weakly, or in the wrong tissue may alter development or physiology dramatically. A stress-response gene expressed transiently may help an organism recover, while the same gene constitutively expressed may be costly or harmful. A regulatory program that stabilizes identity in one context may become pathological if maintained in another.
Biology therefore gains explanatory power when it treats expression as a regulated process unfolding across time, rather than as a static property of genes themselves. Burst kinetics, state transitions, transcriptional memory, RNA half-life, translation rate, protein turnover, chromatin remodeling, and regulatory feedback all shape the realized phenotype. Gene regulation is a temporal control system operating under noise, feedback, environmental change, and historical constraint.
This is especially important for modern single-cell biology. A population of cells may appear homogeneous by tissue label, yet contain many regulatory states, transient intermediates, cycling cells, primed states, stress states, or differentiation trajectories. Gene expression is therefore not only a measurement of “what is on.” It can be a record of state, trajectory, history, and future potential.
Epigenetics, development, and cellular identity
Epigenetics is central to development because development depends on the selective stabilization of different cellular identities from similar or identical genomes. A liver cell, neuron, root cell, pollen grain, immune cell, epithelial cell, or muscle cell does not arise because it contains wholly unique DNA. It arises because particular expression programs are activated, restricted, and maintained through developmental regulation, chromatin organization, signaling, and epigenetic state.
This makes epigenetics one of the strongest explanations for how differentiation becomes durable. Cells acquire identity not only through transient signals, but through regulated patterns of expression that can persist as tissues mature and function. Development is therefore not only a story of genes being present. It is a story of genomes being interpreted differently across space and time.
That is why epigenetics fits so naturally with developmental biology. It helps explain how organisms generate coherent, specialized, long-lived structures from shared hereditary material. Lineage commitment, developmental plasticity, competence windows, reprogramming, tissue maintenance, regeneration, and stem-cell behavior all depend on regulatory memory systems that are neither reducible to raw sequence nor separable from it.
Epigenetics also helps explain why development is sensitive to timing. A signal may have one effect when chromatin is permissive and another when a cell has already passed a competence window. A regulatory pathway may push a progenitor toward one fate early but fail to do so later. Developmental possibility is therefore shaped by both signal and state. Epigenetics is one of the major ways biology studies state.
Epigenetics, physiology, and biological function
Epigenetic regulation matters for physiology because biological function depends on stable yet responsive patterns of activity across tissues and organ systems. Hormonal response, immune activation, stress adaptation, metabolic regulation, tissue repair, circadian organization, neural plasticity, wound response, and inflammatory signaling all depend in part on coordinated changes in gene expression and regulatory state.
This means physiology cannot be understood only as chemistry or organ function in isolation. It is also a problem of regulated genomic activity. Tissues function as they do because they maintain certain expression programs while remaining capable of altering them under changing conditions. Epigenetics provides part of the framework for understanding how this balance is achieved.
Biological function therefore depends not only on what molecules exist, but on whether the right molecular programs can be activated, suppressed, or modulated under the right circumstances. Physiological robustness often depends on regulatory flexibility combined with identity stability. A system must preserve core functional character while remaining responsive to changing internal and external signals.
This balance is visible in immune response, where cells must respond rapidly to pathogens without permanently locking into destructive inflammatory states. It is visible in metabolism, where nutrient availability changes expression programs across tissues. It is visible in neural plasticity, where gene regulation participates in long-term changes in function. Epigenetics therefore helps explain how living systems remain both stable and adaptable.
Ecology, evolution, and sustainability-adjacent biology
Epigenetics is increasingly relevant to ecology and sustainability-adjacent biology because organisms respond to environmental pressures through more than sequence change alone. Drought, salinity, toxins, thermal stress, nutrient limitation, hypoxia, pathogen pressure, acidity, pollution, habitat disruption, and food scarcity can alter gene expression and regulatory states in ways that shape survival, plasticity, development, reproduction, and performance. Environmental change is therefore often mediated through regulatory biology.
This is especially important for sustainability because resilience depends not only on fixed inherited traits, but also on the capacity of living systems to respond adaptively across developmental, physiological, and ecological timescales. Epigenetic responsiveness may shape how populations endure stress, how plants respond to degraded soils, how microbes alter metabolic activity, how marine organisms respond to warming, and how ecosystems recover after disturbance.
Evolutionary biology also intersects with epigenetics because selection acts on organisms whose developmental and physiological states are partly regulated through gene-control systems. Epigenetics does not replace evolution, but it deepens biology’s understanding of how regulated plasticity and inherited architecture shape evolutionary possibility. In practical environmental biology, regulatory responsiveness may influence whether populations persist long enough for longer-term adaptation to matter.
This is not the same as claiming that every environmentally induced expression change is adaptive or heritable. Many changes are transient, costly, pathological, or context-dependent. The sustainability relevance lies in disciplined interpretation: regulation is one pathway through which environmental stress becomes biological response, and that response may influence survival, recovery, vulnerability, or future adaptation.
Marine, freshwater, soil, plant, and microbial relevance
Marine biology makes the environmental relevance of epigenetics especially clear. Marine organisms often face strong gradients in temperature, salinity, oxygen, acidity, light, and nutrient availability, and these conditions can influence gene expression, developmental timing, stress pathways, reproductive output, and physiological state. Coral systems, plankton, fishes, marine invertebrates, macroalgae, seagrasses, and microbial communities all provide contexts in which environmental regulation becomes biologically significant.
Freshwater biology presents similar issues in lakes, rivers, wetlands, and sediments under eutrophication, pollution, warming, fragmentation, hypoxia, and hydrologic change. Regulatory responses can affect development, immune function, stress tolerance, reproduction, and population persistence. In freshwater systems, epigenetic and gene-expression measurements may help reveal biological stress before visible collapse occurs.
Soil biology and microbiology add another important layer because microbial communities often respond rapidly to changes in nutrient supply, redox conditions, toxins, moisture, plant-root exudates, and disturbance through altered regulatory programs. These changes can affect decomposition, nutrient cycling, disease suppression, carbon turnover, and ecosystem recovery. Microbial regulation is especially important because many soil processes are driven by active metabolic state rather than by species presence alone.
Plant science is especially rich in epigenetic relevance. Plants are developmentally plastic organisms that frequently respond to light, drought, temperature, salinity, pathogens, herbivory, and soil conditions through shifts in expression and regulatory state. Agroecology, forestry, and restoration ecology therefore benefit directly from epigenetic thinking because establishment, stress tolerance, regeneration, flowering, seed dormancy, and resilience often depend on regulated plasticity as well as inherited sequence. Across all these domains, gene regulation acts as one of the interfaces through which environment becomes phenotype.
Medical, biomedical, and disease ecology relevance
Epigenetics is foundational to medicine and biomedicine because many diseases involve dysregulated gene expression, abnormal chromatin states, disrupted methylation patterns, altered differentiation, or failures of transcriptional control. Cancer biology is one of the clearest examples, since malignancy often involves both genetic and epigenetic disruption. Developmental disorders, inflammatory states, neurological disease, metabolic disease, immune dysfunction, and aging-related processes also intersect with gene-regulatory biology.
This biomedical relevance matters because disease is not always a matter of damaged sequence alone. A genome may remain largely intact while expression becomes misregulated in ways that alter cell identity, tissue behavior, immune response, or systemic physiology. Epigenetics therefore widened medicine’s explanatory field beyond mutation-only thinking.
Disease ecology adds another scale. Environmental stress, host-pathogen interaction, toxin exposure, chronic inflammation, nutrition, and population-level conditions may alter gene expression and physiological response across communities and species. Epigenetics therefore helps connect biomedical mechanism to ecological context. It is particularly important in understanding plastic response, chronic stress signaling, developmental susceptibility windows, exposure biology, and the layered relationship between environment and phenotype.
Biomedical interpretation also requires ethical and scientific caution. Epigenetic findings can be overinterpreted if they are detached from exposure measurement, tissue specificity, cell composition, developmental timing, confounding variables, and causal evidence. A methylation difference or expression change may be important, but it must be interpreted through rigorous biological design. Epigenetics is powerful precisely because it is contextual; that also makes careless interpretation risky.
Biotechnology, epigenomic analysis, and computational relevance
Biotechnology extends epigenetics into applied systems of analysis, screening, engineering, and intervention. Epigenomic profiling, chromatin accessibility assays, methylation analysis, single-cell expression profiling, stem-cell differentiation studies, tissue engineering, crop regulation studies, microbial engineering, environmental stress-response assays, and drug-screening workflows all depend on the ability to measure gene-regulatory state at scale.
Computational biology is especially important here because epigenetics generates complex, high-dimensional data. Chromatin-state maps, methylation matrices, transcriptomic datasets, accessibility profiles, lineage trajectories, and cell-state transitions cannot be interpreted well without statistical and computational methods. Epigenetics is therefore one of the clearest places where molecular biology becomes systems biology and data science at once.
This also makes epigenetics a strong bridge between basic and applied biology. The same principles that explain how cell identity is maintained can inform regenerative medicine, crop improvement, microbial engineering, environmental response analysis, toxicity screening, disease classification, and biotechnology design. In practical terms, computational epigenetics now includes normalization, differential expression, accessibility scoring, trajectory inference, clustering, regulatory-network inference, multi-omics integration, and model-based reconstruction of regulatory state spaces.
Reproducibility is especially important in epigenomic work. A regulatory claim may depend on sequencing depth, peak calling, methylation thresholds, batch correction, cell-type composition, normalization, reference genome choice, tissue handling, and environmental metadata. The strongest computational epigenetics therefore pairs statistical sophistication with transparent provenance and biological validation.
Quantitative epigenetics: mathematics, R, and Python
Epigenetics is not only mechanistic. It is also quantitative. Transcript abundance, state transitions, methylation fractions, chromatin accessibility, regulatory decay, regulatory activation, expression change, and regulatory concordance can all be modeled mathematically and analyzed computationally. This does not reduce regulation to numbers, but it makes regulatory dynamics more explicit, testable, and reproducible.
A simple model for transcript decline is:
Interpretation: Transcript abundance declines exponentially when degradation proceeds at an approximately constant proportional rate.
where \(m(t)\) is transcript abundance at time \(t\), \(m_0\) is initial abundance, and \(k\) is the decay constant. This is useful because gene expression depends not only on transcriptional activation, but also on RNA stability and turnover.
The associated half-life is:
Interpretation: Transcript half-life is the time required for RNA abundance to decline by half.
This converts a fitted decay constant into a biologically interpretable timescale.
A more useful continuous model for transcript dynamics includes both production and decay:
Interpretation: Transcript abundance reflects a balance between time-dependent production and degradation.
where \(\alpha(t)\) is the time-dependent transcription or effective production rate and \(\beta\) is the degradation constant. This matters because expression is often governed by changing input rather than decay alone.
A simple expression-state model can treat a regulatory locus as switching between inactive and active states with transition rates \(k_{\mathrm{on}}\) and \(k_{\mathrm{off}}\):
Interpretation: The probability that a regulatory state is active changes according to activation and inactivation rates.
While simplified, this captures an important truth: epigenetic regulation often shapes the probability and persistence of transcriptional activity rather than determining one fixed absolute output.
If \(M\) is the number of methylated reads or sites and \(U\) is the unmethylated count, a simple methylation fraction is:
Interpretation: Methylation fraction estimates the share of observations that are methylated at a locus or region.
This is useful because many epigenomic assays produce proportion-based measurements rather than binary on/off states.
A simple accessibility change can be written as:
Interpretation: Accessibility change measures how much chromatin accessibility differs between two conditions.
A simple expression fold change can be written as:
Interpretation: Log2 fold change expresses expression differences on a symmetric scale.
where \(A\) is accessibility, \(E\) is expression, and \(\epsilon\) is a small pseudocount. Concordance between \(\Delta A\) and \(\log_2FC\) can help screen whether chromatin opening and expression change move in the same direction.
Variables, units, and epigenetic interpretation
Quantitative epigenetics depends on variables that connect transcript abundance, RNA decay, regulatory state, methylation, chromatin accessibility, expression change, and biological interpretation. The table below summarizes several central quantities.
| Symbol or Term | Meaning | Typical Unit or Scale | Epigenetic Interpretation |
|---|---|---|---|
| \(m(t)\) | Transcript abundance at time \(t\) | counts, TPM, normalized expression, fluorescence, or arbitrary units | Amount of RNA transcript measured or modeled at a given time |
| \(m_0\) | Initial transcript abundance | same as \(m(t)\) | Starting transcript level before decay, perturbation, or regulatory change |
| \(k\) | Decay constant | per unit time | Rate of exponential transcript loss |
| \(t_{1/2}\) | Transcript half-life | time | Time required for transcript abundance to decline by half |
| \(\alpha(t)\) | Production rate | abundance per time | Time-dependent transcriptional input or effective expression production |
| \(\beta\) | Degradation constant | per unit time | Rate of transcript removal in production-decay models |
| \(P_{\mathrm{on}}\) | Probability or fraction of active regulatory state | fraction from 0 to 1 | Share of cells, loci, or modeled state probability in active condition |
| \(k_{\mathrm{on}}\) | Activation transition rate | per unit time | Rate at which inactive loci or states become active |
| \(k_{\mathrm{off}}\) | Inactivation transition rate | per unit time | Rate at which active loci or states become inactive |
| \(M\) | Methylated count | reads, sites, molecules, or observations | Number of observations classified as methylated |
| \(U\) | Unmethylated count | reads, sites, molecules, or observations | Number of observations classified as unmethylated |
| \(f_{\mathrm{meth}}\) | Methylation fraction | fraction from 0 to 1 | Proportion of methylated observations at a locus, region, or sample |
| \(A_{\mathrm{control}}, A_{\mathrm{treated}}\) | Chromatin accessibility in two conditions | normalized signal, peak score, read density, or assay units | Relative openness of chromatin under comparison conditions |
| \(\Delta A\) | Accessibility change | difference in accessibility units | Direction and magnitude of chromatin accessibility shift |
| \(E_{\mathrm{control}}, E_{\mathrm{treated}}\) | Expression values in two conditions | counts, TPM, normalized counts, or assay units | Gene-expression levels being compared across conditions |
| \(\epsilon\) | Pseudocount | same as expression value | Small value used to avoid division by zero in fold-change calculations |
| \(\log_2FC\) | Log2 fold change | log2 ratio | Symmetric expression-change measure across conditions |
| Concordance | Agreement between regulatory layers | classification or correlation | Whether accessibility, methylation, and expression move in compatible directions |
The table shows why epigenetic quantities require context. A methylation fraction, accessibility score, expression fold change, or state-transition rate becomes biologically meaningful only when linked to cell type, tissue, developmental stage, assay method, environmental condition, normalization procedure, and biological question.
Worked example: transcript half-life, methylation, and regulatory concordance
Suppose a transcript starts at m0 = 120 arbitrary units and declines to 30 units after 6 hours. Under the exponential decay model:
Interpretation: Transcript abundance declines according to initial abundance, decay constant, and elapsed time.
Substituting:
Interpretation: The observed decline can be used to estimate the transcript decay constant.
Dividing both sides:
Interpretation: The transcript has declined to 25 percent of its initial abundance.
Taking the natural logarithm:
Interpretation: Log transformation converts the exponential relation into a solvable linear expression.
Solving:
Interpretation: The estimated transcript decay constant is approximately 0.2310 per hour.
The half-life is:
Interpretation: The transcript half-life is approximately 3 hours under this simplified model.
This is useful because it converts expression decline into a biologically interpretable dynamic parameter.
Now suppose a locus has M = 72 methylated observations and U = 28 unmethylated observations. Its methylation fraction is:
Interpretation: The locus is methylated in 72 percent of observed reads, sites, or molecules under this simplified measurement.
This is useful because epigenomic assays often require proportion-based interpretation rather than simple binary labels.
Finally, suppose chromatin accessibility rises from Acontrol = 0.35 to Atreated = 0.62, while expression rises from Econtrol = 20 to Etreated = 80. Accessibility change is:
Interpretation: Chromatin accessibility increased by 0.27 normalized units.
Expression change is:
Interpretation: Expression increased fourfold, corresponding to a log2 fold change of 2.
This is a concordant regulatory pattern because accessibility and expression both increased. In real epigenomic analysis, concordance does not prove causation by itself, but it helps prioritize loci for deeper investigation.
Computational modeling
Computational modeling helps make epigenetics explicit because regulation is dynamic, probabilistic, layered, and context-dependent. Transcript-decay models estimate RNA stability. Production-decay models represent expression under changing transcriptional input. Two-state switching models approximate active and inactive regulatory states. Methylation summaries quantify locus-level or region-level epigenomic state. Accessibility scores summarize chromatin openness. Differential expression identifies regulated genes. Concordance screens compare whether expression and accessibility move together. Markov-style transition matrices provide a simplified way to represent cell-state progression and regulatory memory.
The selected examples below focus on compact, reusable workflows: transcript half-life estimation, production-decay simulation, regulatory-state switching, methylation fraction calculation, differential expression, accessibility change, regulatory concordance classification, and Markov-style cell-state transitions. The GitHub repository extends the same logic into richer workflows for SQL provenance, reproducible data files, validation notes, notebooks, epigenetic condition scoring, and multi-language scientific-computing examples.
The purpose is not to reduce epigenetics to code. The purpose is to make regulatory reasoning inspectable. An epigenetic claim becomes stronger when expression data, methylation measurements, accessibility scores, state definitions, sample metadata, assay details, normalization choices, and analytical code are documented together.
R workflow: expression decay, regulatory switching, methylation, and accessibility
R is useful for epigenetics because it supports statistical fitting, time-series summaries, tabular data analysis, assay integration, and reproducible reporting. The following workflow estimates expression decay and half-life, simulates production-decay dynamics, models regulatory-state switching, summarizes methylation fractions, and integrates expression with chromatin accessibility.
# Epigenetics, Regulation, and Gene Expression Workflow
#
# This workflow demonstrates five quantitative epigenetics tasks:
#
# 1. Fit simple transcript decay and estimate half-life.
# 2. Simulate production-decay expression dynamics.
# 3. Simulate two-state regulatory switching.
# 4. Summarize methylation fractions.
# 5. Integrate expression and chromatin-accessibility change.
#
# These examples can be adapted for molecular biology, developmental biology,
# environmental stress response, single-cell biology, epigenomic profiling,
# plant science, disease biology, biotechnology, and computational biology.
library(dplyr)
library(tidyr)
library(tibble)
# ------------------------------------------------------------
# 1. Expression decay and half-life estimation
# ------------------------------------------------------------
decay_df <- tibble(
time_h = c(0, 1, 2, 4, 6, 8),
expr = c(120, 98, 76, 49, 31, 20)
)
decay_fit <- lm(log(expr) ~ time_h, data = decay_df)
k_est <- -coef(decay_fit)[["time_h"]]
m0_est <- exp(coef(decay_fit)[["(Intercept)"]])
half_life_h <- log(2) / k_est
decay_summary <- tibble(
k_est = k_est,
m0_est = m0_est,
half_life_h = half_life_h,
r_squared_log_space = summary(decay_fit)$r.squared
)
decay_df <- decay_df %>%
mutate(
pred_expr = exp(predict(decay_fit)),
residual = expr - pred_expr
)
# ------------------------------------------------------------
# 2. Production-decay expression dynamics
# ------------------------------------------------------------
simulate_prod_decay <- function(
times,
alpha_base = 2,
alpha_pulse = 18,
pulse_start = 3,
pulse_end = 8,
beta = 0.35,
m0 = 5
) {
m <- numeric(length(times))
m[1] <- m0
for (i in 2:length(times)) {
dt <- times[i] - times[i - 1]
alpha_t <- ifelse(
times[i - 1] >= pulse_start & times[i - 1] <= pulse_end,
alpha_pulse,
alpha_base
)
dm <- alpha_t - beta * m[i - 1]
m[i] <- max(m[i - 1] + dm * dt, 0)
}
tibble(time_h = times, expression = m)
}
prod_decay_df <- simulate_prod_decay(seq(0, 20, by = 0.1))
# ------------------------------------------------------------
# 3. Two-state regulatory switching
# ------------------------------------------------------------
simulate_state_switch <- function(
times,
kon = 0.35,
koff = 0.08,
p_on0 = 0.05
) {
p_on <- numeric(length(times))
p_on[1] <- p_on0
for (i in 2:length(times)) {
dt <- times[i] - times[i - 1]
dp <- kon * (1 - p_on[i - 1]) - koff * p_on[i - 1]
p_on[i] <- min(max(p_on[i - 1] + dp * dt, 0), 1)
}
tibble(
time_h = times,
p_on = p_on,
p_off = 1 - p_on
)
}
state_df <- simulate_state_switch(seq(0, 25, by = 0.1))
# ------------------------------------------------------------
# 4. Methylation fraction summary
# ------------------------------------------------------------
meth_df <- tibble(
locus = paste0("locus_", 1:8),
methylated = c(85, 20, 61, 42, 95, 14, 50, 73),
unmethylated = c(15, 80, 39, 58, 5, 86, 50, 27)
) %>%
mutate(
total_observations = methylated + unmethylated,
meth_fraction = methylated / total_observations
)
meth_summary <- meth_df %>%
summarise(
mean_methylation = mean(meth_fraction),
median_methylation = median(meth_fraction),
high_methylation_loci = sum(meth_fraction >= 0.70),
low_methylation_loci = sum(meth_fraction <= 0.30)
)
# ------------------------------------------------------------
# 5. Integrated expression and accessibility summary
# ------------------------------------------------------------
reg_df <- tibble(
gene = c("GATA3", "HSP70", "DNMT1", "MYC", "COL1A1", "NRF2"),
control_expr = c(12.0, 18.0, 22.0, 45.0, 31.0, 14.0),
treated_expr = c(25.0, 60.0, 17.0, 30.0, 20.0, 29.0),
control_access = c(0.32, 0.41, 0.55, 0.71, 0.48, 0.36),
treated_access = c(0.56, 0.73, 0.46, 0.49, 0.33, 0.61)
) %>%
mutate(
log2FC_expr = log2((treated_expr + 1e-6) / (control_expr + 1e-6)),
delta_access = treated_access - control_access,
regulatory_pattern = case_when(
log2FC_expr > 0 & delta_access > 0 ~ "up_with_opening",
log2FC_expr < 0 & delta_access < 0 ~ "down_with_closing",
TRUE ~ "discordant_or_complex"
)
)
pattern_summary <- reg_df %>%
count(regulatory_pattern)
print(round(decay_summary, 4))
print(decay_df %>% mutate(across(where(is.numeric), round, 4)))
print(prod_decay_df %>% slice_head(n = 12) %>% mutate(expression = round(expression, 4)))
print(prod_decay_df %>% slice_tail(n = 12) %>% mutate(expression = round(expression, 4)))
print(state_df %>% slice_head(n = 12) %>% mutate(across(where(is.numeric), round, 4)))
print(state_df %>% slice_tail(n = 12) %>% mutate(across(where(is.numeric), round, 4)))
print(meth_df %>% mutate(meth_fraction = round(meth_fraction, 4)))
print(round(meth_summary, 4))
print(reg_df %>% arrange(desc(log2FC_expr)) %>% mutate(across(where(is.numeric), round, 4)))
print(pattern_summary)This R workflow is useful because epigenomic interpretation often requires more than a single expression number. A strong regulatory analysis often integrates RNA dynamics, regulatory-state persistence, methylation proportions, chromatin accessibility, and expression change.
Python workflow: regulatory-state dynamics, expression output, accessibility, and cell-state transitions
Python is useful for epigenetics because it supports numerical simulation, matrix operations, time-series analysis, assay integration, tabular screening, and reproducible computation. The following workflow estimates expression half-life, simulates regulatory-state dynamics coupled to expression output, summarizes differential expression and accessibility concordance, and models Markov-style cell-state transitions.
"""
Epigenetics, Regulation, and Gene Expression Workflow
This workflow demonstrates four quantitative epigenetics tasks:
1. Estimate expression decay, transcript half-life, and area under the curve.
2. Simulate regulatory-state dynamics coupled to expression output.
3. Summarize differential expression and chromatin accessibility concordance.
4. Model Markov-style cell-state transitions.
The examples are compact, but the same structures can be extended to
molecular biology, developmental biology, environmental stress response,
single-cell biology, epigenomic profiling, plant science, disease biology,
biotechnology, and computational biology.
"""
from __future__ import annotations
import numpy as np
import pandas as pd
def expression_half_life_example() -> tuple[pd.DataFrame, pd.DataFrame]:
"""
Estimate transcript half-life and area under the expression curve.
"""
time_h = np.array([0, 1, 2, 4, 6, 8], dtype=float)
expr = np.array([120, 98, 76, 49, 31, 20], dtype=float)
slope, intercept = np.polyfit(time_h, np.log(expr), 1)
k_est = -slope
m0_est = np.exp(intercept)
half_life_h = np.log(2.0) / k_est
auc = np.trapz(expr, time_h)
predicted = np.exp(intercept + slope * time_h)
summary = pd.DataFrame(
{
"k_est": [k_est],
"m0_est": [m0_est],
"half_life_h": [half_life_h],
"AUC": [auc],
}
)
trace = pd.DataFrame(
{
"time_h": time_h,
"expr_observed": expr,
"expr_predicted": predicted,
"residual": expr - predicted,
}
)
return summary, trace
def simulate_regulatory_expression(
t_end: float = 30.0,
dt: float = 0.05,
kon: float = 0.28,
koff: float = 0.10,
alpha_on: float = 14.0,
alpha_off: float = 1.0,
beta: float = 0.35,
p_on0: float = 0.05,
m0: float = 2.0,
) -> pd.DataFrame:
"""
Simulate regulatory activation probability and expression output.
"""
times = np.arange(0.0, t_end + dt, dt)
p_on = np.zeros_like(times)
expression = np.zeros_like(times)
p_on[0] = p_on0
expression[0] = m0
for i in range(1, len(times)):
dp = kon * (1.0 - p_on[i - 1]) - koff * p_on[i - 1]
p_on[i] = np.clip(p_on[i - 1] + dp * dt, 0.0, 1.0)
alpha_t = alpha_on * p_on[i] + alpha_off * (1.0 - p_on[i])
d_expression = alpha_t - beta * expression[i - 1]
expression[i] = max(expression[i - 1] + d_expression * dt, 0.0)
return pd.DataFrame(
{
"time": times,
"p_on": p_on,
"expression": expression,
}
)
def differential_expression_accessibility_summary() -> tuple[pd.DataFrame, pd.DataFrame]:
"""
Summarize expression and accessibility concordance.
"""
data = pd.DataFrame(
{
"gene": ["GATA3", "HSP70", "DNMT1", "MYC", "COL1A1", "NRF2"],
"control_expr": [12.0, 18.0, 22.0, 45.0, 31.0, 14.0],
"treated_expr": [25.0, 60.0, 17.0, 30.0, 20.0, 29.0],
"control_access": [0.32, 0.41, 0.55, 0.71, 0.48, 0.36],
"treated_access": [0.56, 0.73, 0.46, 0.49, 0.33, 0.61],
}
)
data["log2FC_expr"] = np.log2(
(data["treated_expr"] + 1e-6)
/ (data["control_expr"] + 1e-6)
)
data["delta_access"] = data["treated_access"] - data["control_access"]
def classify(row: pd.Series) -> str:
if row["log2FC_expr"] > 0 and row["delta_access"] > 0:
return "up_with_opening"
if row["log2FC_expr"] < 0 and row["delta_access"] < 0:
return "down_with_closing"
return "discordant_or_complex"
data["regulatory_pattern"] = data.apply(classify, axis=1)
summary = (
data.groupby("regulatory_pattern")
.size()
.reset_index(name="n_genes")
.sort_values("n_genes", ascending=False)
)
return data.sort_values("log2FC_expr", ascending=False), summary
def methylation_summary() -> tuple[pd.DataFrame, pd.DataFrame]:
"""
Summarize methylation fractions for a small epigenomic table.
"""
methylation = pd.DataFrame(
{
"locus": [f"locus_{i}" for i in range(1, 9)],
"methylated": [85, 20, 61, 42, 95, 14, 50, 73],
"unmethylated": [15, 80, 39, 58, 5, 86, 50, 27],
}
)
methylation["total_observations"] = (
methylation["methylated"] + methylation["unmethylated"]
)
methylation["meth_fraction"] = (
methylation["methylated"] / methylation["total_observations"]
)
summary = pd.DataFrame(
{
"mean_methylation": [methylation["meth_fraction"].mean()],
"median_methylation": [methylation["meth_fraction"].median()],
"high_methylation_loci": [np.sum(methylation["meth_fraction"] >= 0.70)],
"low_methylation_loci": [np.sum(methylation["meth_fraction"] <= 0.30)],
}
)
return methylation, summary
def markov_cell_state_transition() -> pd.DataFrame:
"""
Simulate a Markov-style cell-state transition matrix.
"""
states = ["stem_like", "primed", "differentiated"]
transition_matrix = np.array(
[
[0.78, 0.20, 0.02],
[0.05, 0.80, 0.15],
[0.00, 0.08, 0.92],
]
)
state_vector = np.array([0.85, 0.10, 0.05])
trajectory = [state_vector.copy()]
for _ in range(15):
state_vector = state_vector @ transition_matrix
trajectory.append(state_vector.copy())
trajectory_df = pd.DataFrame(trajectory, columns=states)
trajectory_df["step"] = range(len(trajectory_df))
return trajectory_df
def main() -> None:
"""
Run compact epigenetics and gene-expression workflows.
"""
half_life_summary, half_life_trace = expression_half_life_example()
regulatory_df = simulate_regulatory_expression()
expression_access_df, expression_access_summary = differential_expression_accessibility_summary()
methylation_df, methylation_summary_df = methylation_summary()
cell_state_df = markov_cell_state_transition()
print("Expression half-life summary:")
print(half_life_summary.round(4).to_string(index=False))
print(half_life_trace.round(4).to_string(index=False))
print("\nRegulatory-state expression dynamics:")
print(regulatory_df.head(15).round(4).to_string(index=False))
print(regulatory_df.tail(15).round(4).to_string(index=False))
print("\nExpression and accessibility concordance:")
print(expression_access_df.round(4).to_string(index=False))
print(expression_access_summary.to_string(index=False))
print("\nMethylation fractions:")
print(methylation_df.round(4).to_string(index=False))
print(methylation_summary_df.round(4).to_string(index=False))
print("\nMarkov-style cell-state trajectory:")
print(cell_state_df.round(4).to_string(index=False))
if __name__ == "__main__":
main()This Python workflow is useful because epigenetic regulation often requires linking state probability, expression output, chromatin accessibility, methylation fraction, and cell-state transition in one reproducible scaffold. It provides a practical bridge between molecular regulation, single-cell thinking, assay integration, and computational systems biology.
GitHub repository
The article body includes compact R and Python examples so the biological and scientific argument remains readable. The full repository expands those examples into a broader computational epigenetics and gene-expression workflow, including expression-decay fitting, production-decay modeling, two-state regulatory switching, methylation-fraction summaries, differential expression, accessibility scoring, regulatory concordance screening, Markov-style cell-state transitions, epigenetic condition scoring, SQL provenance structures, reproducible data files, validation notes, and full-stack scientific-computing examples across Python, R, Julia, Fortran, Rust, Go, C, C++, SQL, and notebooks.
Complete Code Repository
The full code distribution for this article, including selected article examples, expanded computational workflows, reproducible data structures, provenance documentation, validation notes, and full-stack scientific-computing scaffolding, is available on GitHub.
Limits, complexity, and modern epigenetic thinking
Epigenetics is powerful, but it is also a field where precision matters. Not every persistent change in expression should be called epigenetic in the strongest sense, and not every chromatin-associated correlation is a causal regulatory mechanism. Biology is strongest here when it distinguishes clearly among sequence effects, chromatin states, transcriptional outcomes, developmental memory, environmentally induced regulatory changes, technical artifacts, and cell-composition effects.
This is why modern epigenetic thinking increasingly emphasizes integration rather than exaggeration. Epigenetics does not replace genetics, development, physiology, ecology, or evolution. It helps explain how these domains are linked through regulatory architecture. The field is most useful when it remains connected to careful molecular, cellular, organismal, ecological, and computational biology.
Models are useful because they clarify assumptions, expose mechanisms, and make comparison possible. But a methylation fraction is not a whole regulatory state, a transcript-decay curve is not a complete gene-control system, and an accessibility score is not direct proof of causation. Quantitative tools are strongest when they support biological interpretation rather than replacing it.
In that sense, epigenetics provides a model case of modern biology itself: mechanistically rich, developmentally important, environmentally responsive, quantitatively analyzable, and irreducible to one single level of explanation. Its strength lies in showing how control, context, and history shape the meaning of genomic information.
This caution is especially important because epigenetic claims can easily be overstated in public discourse. Environmental effects on gene expression are real, but that does not mean every experience leaves a stable inheritable mark. Epigenetic inheritance, developmental memory, cellular regulation, and transient expression response are related but distinct phenomena. Scientific rigor requires keeping those distinctions visible.
Why this matters for scientific work
For working scientists, epigenetics matters because many biological questions are misread when regulation is treated as a black box between gene and phenotype. A developmental problem may depend on chromatin accessibility rather than gene absence. A physiological problem may depend on altered transcriptional timing rather than structural tissue damage. A restoration or crop problem may depend on stress-responsive regulation rather than constitutive trait difference alone. A disease problem may involve durable dysregulation without dramatic sequence change.
This means epigenetics should often be treated as explanatory infrastructure rather than as a specialty topic detached from the rest of biology. Developmental biologists need it because cellular identity depends on regulatory memory. Physiologists need it because tissues operate through controlled expression programs. Ecologists need it because organisms respond to stress through regulatory systems. Plant scientists need it because development and environmental plasticity are strongly regulated. Medical researchers need it because dysregulation is a major mechanism of disease. Computational biologists need it because epigenomic data are central to modern biological inference.
The scientific importance of epigenetics lies partly in this breadth. It is one of the principal ways biology explains how living systems interpret their own genomes under real conditions of development, physiology, disease, and environment.
Epigenetics is also practically actionable. Transcript half-lives can be estimated. Methylation fractions can be measured. Accessibility profiles can be compared. Regulatory concordance can be screened. Cell-state transitions can be modeled. These tools connect regulatory biology to medicine, crop science, restoration ecology, environmental monitoring, biotechnology, single-cell analysis, and systems biology.
Conclusion
Epigenetics, regulation, and gene expression show that biological information is not only stored in DNA sequence, but also regulated through structured systems of accessibility, modification, transcriptional control, RNA dynamics, and cellular memory. These systems help explain how similar genomes generate different tissues, how organisms respond to environments, how biological states persist, and how disease can arise through dysregulated control rather than sequence change alone.
To understand epigenetics is therefore to understand one of the deepest conditions of biological organization. Gene activity is not merely present or absent. It is regulated, contextual, layered, dynamic, and historically conditioned. That is why epigenetics remains central not only to molecular biology and development, but also to physiology, ecology, plant science, marine and freshwater biology, microbiology, disease ecology, medicine, biotechnology, and sustainability-adjacent biology more broadly.
Epigenetics is thus more than a refinement of genetics. It is one of the principal ways biology explains how living systems interpret their own genomes. Modern quantitative and computational workflows deepen that understanding by making transcript dynamics, methylation, accessibility, regulatory-state transitions, and epigenomic provenance more transparent, reproducible, and scientifically interpretable.
Related articles
- Biology
- DNA, RNA, and the Molecular Logic of Life
- Molecular Biology and the Flow of Genetic Information
- Cell Signaling, Communication, and Biological Coordination
- Development, Differentiation, and the Making of Organisms
- Genes, Inheritance, and the Principles of Heredity
- Genomics and the Expansion of Biological Knowledge
- Physiology and the Regulation of Living Systems
- Systems Biology and the Logic of Biological Integration
- Evolution and the History of Life
- Microbiology and the Hidden Majority of Life
Further reading
- Alberts, B. et al. (2002) Molecular Biology of the Cell. 4th edn. New York: Garland Science. NCBI Bookshelf edition available at: https://www.ncbi.nlm.nih.gov/books/NBK21054/
- Alberts, B. et al. (2002) ‘Chromosomal DNA and its packaging in the chromatin fiber’, in Molecular Biology of the Cell. 4th edn. Available at: https://www.ncbi.nlm.nih.gov/books/NBK26834/
- Alberts, B. et al. (2002) ‘From DNA to RNA’, in Molecular Biology of the Cell. 4th edn. Available at: https://www.ncbi.nlm.nih.gov/books/NBK26887/
- Allis, C.D., Jenuwein, T. and Reinberg, D. (eds.) (2007) Epigenetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. Publisher information available at: https://www.cshlpress.com/default.tpl?action=full&cart=174569904910328318&type=full&–eqskudatarq=450
- ENCODE Project Consortium (2012) ‘An integrated encyclopedia of DNA elements in the human genome’, Nature, 489, pp. 57–74. Available at: https://doi.org/10.1038/nature11247
- ENCODE Project Consortium et al. (2020) ‘Expanded encyclopaedias of DNA elements in the human and mouse genomes’, Nature, 583, pp. 699–710. Available at: https://doi.org/10.1038/s41586-020-2493-4
- Gilbert, S.F. (2000) Developmental Biology. 6th edn. Sunderland, MA: Sinauer Associates. NCBI Bookshelf edition available at: https://www.ncbi.nlm.nih.gov/books/NBK9983/
- National Human Genome Research Institute (2023) The Encyclopedia of DNA Elements (ENCODE). Available at: https://www.genome.gov/Funded-Programs-Projects/ENCODE-Project-ENCyclopedia-Of-DNA-Elements
- Ruskin, H.J., O’Connor, C. and Shelton, D.N. (2017) ‘Recent advances in computational epigenetics’, Advances in Genomics and Genetics, 7, pp. 1–12. Available at: https://doi.org/10.2147/AGG.S115524
- Zhang, Y. et al. (2016) ‘Jointly characterizing epigenetic dynamics across multiple human cell types’, Nucleic Acids Research, 44(14), pp. 6721–6731. Available at: https://doi.org/10.1093/nar/gkw278
References
- Alberts, B. et al. (2002) Molecular Biology of the Cell. 4th edn. New York: Garland Science. NCBI Bookshelf edition available at: https://www.ncbi.nlm.nih.gov/books/NBK21054/
- Alberts, B. et al. (2002) ‘Chromosomal DNA and its packaging in the chromatin fiber’, in Molecular Biology of the Cell. 4th edn. Available at: https://www.ncbi.nlm.nih.gov/books/NBK26834/
- Alberts, B. et al. (2002) ‘From DNA to RNA’, in Molecular Biology of the Cell. 4th edn. Available at: https://www.ncbi.nlm.nih.gov/books/NBK26887/
- Allis, C.D., Jenuwein, T. and Reinberg, D. (eds.) (2007) Epigenetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. Publisher information available at: https://www.cshlpress.com/default.tpl?action=full&cart=174569904910328318&type=full&–eqskudatarq=450
- ENCODE Project Consortium (2012) ‘An integrated encyclopedia of DNA elements in the human genome’, Nature, 489, pp. 57–74. Available at: https://doi.org/10.1038/nature11247
- ENCODE Project Consortium et al. (2020) ‘Expanded encyclopaedias of DNA elements in the human and mouse genomes’, Nature, 583, pp. 699–710. Available at: https://doi.org/10.1038/s41586-020-2493-4
- Gilbert, S.F. (2000) Developmental Biology. 6th edn. Sunderland, MA: Sinauer Associates. NCBI Bookshelf edition available at: https://www.ncbi.nlm.nih.gov/books/NBK9983/
- National Human Genome Research Institute (2023) The Encyclopedia of DNA Elements (ENCODE). Available at: https://www.genome.gov/Funded-Programs-Projects/ENCODE-Project-ENCyclopedia-Of-DNA-Elements
- Ruskin, H.J., O’Connor, C. and Shelton, D.N. (2017) ‘Recent advances in computational epigenetics’, Advances in Genomics and Genetics, 7, pp. 1–12. Available at: https://doi.org/10.2147/AGG.S115524
- Zhang, Y. et al. (2016) ‘Jointly characterizing epigenetic dynamics across multiple human cell types’, Nucleic Acids Research, 44(14), pp. 6721–6731. Available at: https://doi.org/10.1093/nar/gkw278